





Abstract
The Consensus Coding Sequence (CCDS) collaboration involves curators at multiple centers with a goal of producing a conservative set of high quality, protein-coding region annotations for the human and mouse reference genome assemblies. The CCDS data set reflects a ‘gold standard’ definition of best supported protein annotations, and corresponding genes, which pass a standard series of quality assurance checks and are supported by manual curation. This data set supports use of genome annotation information by human and mouse researchers for effective experimental design, analysis and interpretation. The CCDS project consists of analysis of automated whole-genome annotation builds to identify identical CDS annotations, quality assurance testing and manual curation support. Identical CDS annotations are tracked with a CCDS identifier (ID) and any future change to the annotated CDS structure must be agreed upon by the collaborating members. CCDS curation guidelines were developed to address some aspects of curation in order to improve initial annotation consistency and to reduce time spent in discussing proposed annotation updates. Here, we present the current status of the CCDS database and details on our procedures to track and coordinate our efforts. We also present the relevant background and reasoning behind the curation standards that we have developed for CCDS database treatment of transcripts that are nonsense-mediated decay (NMD) candidates, for transcripts containing upstream open reading frames, for identifying the most likely translation start codons and for the annotation of readthrough transcripts. Examples are provided to illustrate the application of these guidelines.
Database URL: http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi
Introduction
One of the fundamental aspects of an organism’s genome is its genes, which provide the instructions for the production of both mRNAs that encode proteins and functional noncoding RNAs involved in regulation of gene expression and protein-coding gene translation. Many researchers rely on human and mouse genome annotation for the design and interpretation of experiments. Several such data sets exist produced by both manual and automated protocols that may employ different methods and standards for annotation, as well as different sources of input sequence data. There is often a trade-off between coverage and accuracy and therefore one must decide how conservative a set to use. However, it is difficult to compare methods and protocols used by different annotation groups and pipelines, and challenging to determine if different genome annotation browsers are displaying identical annotation information or alternate interpretations of the underlying primary data. It is important to be aware of alternate annotation information in order to choose the most appropriate data set for further analysis or experimental design.
In order to address this issue, the Consensus Coding Sequence (CCDS) project (1) provides a conservative set of consensus protein-coding sequences for human and mouse. The nature of the project promotes collaboration between annotation groups from different institutions—the National Center for Biotechnology Information (NCBI), the Wellcome Trust Sanger Institute (WTSI), the European Bioinformatics Institute (EBI) and the University of California Santa Cruz (UCSC). All groups contribute in different ways to the project: three groups provide the genome annotation data sets (EBI, WTSI and NCBI); three groups provide curation review (NCBI, WTSI and UCSC) and two groups support quality assurance tests for the CCDS data set (NCBI and UCSC). Ensembl annotations (2), a joint project between WTSI and EBI, include both automated predictions from a computational annotation process and manual annotations from the Human and Vertebrate Analysis and Annotation (HAVANA) group (3, 4) (WTSI), whereas the NCBI annotations include RefSeq records (5, 6) produced from both manual and automated annotations. Thus, curation support is an integral factor supporting the NCBI and Ensembl genome annotation data sets, and for the CCDS data set. The high confidence CCDS reference set is built based on comparison of NCBI and Ensembl genome annotation data to identify identical CDS genomic coordinates (same start and stop codons, same splice site coordinates). The annotation must also pass a number of stringent quality assurance tests. The CCDS data set is provided as periodic species-specific comprehensive releases; however, there is also an ongoing coordinated review process that contributes annotation modifications and additions to the NCBI and Ensembl genome annotation data set and to the CCDS build process. It is important to note that manual curation supports the data set but not all CCDS IDs have been curated, and that the CCDS project is conservative which is both a strength and weakness. For example, some known genes are excluded from the data set when the assembled genome sequence cannot support annotating the correct CDS, and some consistently annotated CDS regions are intentionally removed from the CCDS data set if there are quality concerns (as described below).
Status of the CCDS data set
CCDS builds occur whenever the human or mouse genomes are re-annotated by NCBI, coupled with timing considerations for Ensembl data set releases. Re-annotation occurs at irregular intervals to update annotation on the same assembly, or when a new genome assembly is released. The most recent CCDS comparative analysis was on the human genome with results released on 7 September 2011. Compared with the previous annotation comparison analysis (released in April 2011), the human CCDS data set increased by 909 CCDS IDs, which includes adding additional protein isoforms for genes that were already represented in the data set with at least one CCDS ID, as well as adding representation for 64 Gene IDs that were not previously included (Table 1). Mouse has 137 more genes with a CCDS ID than human but has fewer CCDS IDs overall, so fewer alternative splice variants are included in the mouse CCDS data set. This is due to more focused curation on human genome annotation than on mouse.
Status of current CCDS builds (as of 7 September 2011)
| Organism → | Human (Build 37.3) | Mouse (Build 37.2) |
|---|---|---|
| GeneIDs | 18 471 | 19 508 |
| CCDS IDs | 26 473 | 22 187 |
| Public CCDS IDsa | 26 400 | 21 921 |
| Genes with >1 CCDS ID | 4999 | 1986 |
| Genes with >6 CCDS IDs | 76 | 15 |
aPublic CCDS IDs are all those that are not currently under review or pending an update or withdrawal
The CCDS data set size continues to increase with each analysis based on both the computational genome annotation updates, which integrate new data sets submitted to the International Nucleotide Sequence Database Collaboration (INSDC, http://www.insdc.org/), and on ongoing curation activities that supplement or improve upon that annotation.
Quality assurance testing for the CCDS database
In order to ensure that consensus coding sequences are of high quality, multiple quality assurance (QA) tests are done. While each of the collaborating groups independently performs QA tests in their annotation pipelines, an additional layer of QA tests are applied to the set of identified matching genome annotations prior to accepting them and assigning a CCDS ID (Table 2). Candidate annotations that fail these QA tests undergo a round of manual checking, which provides feedback that may be useful to the NCBI and Ensembl automatic annotation pipelines, may result in improved QA tests and may result in a curatorial decision to reject annotation matches based on the QA failure or rescue a match based on biological knowledge of an exception category. For example, manual review of QA results for earlier analysis runs resulted in a QA override decision for selenocysteine proteins which were flagged with an error for internal stop codons. These tests continue to identify some CDS annotations added by automatic processes based on new primary data submissions that likely do not represent bona fide coding sequences; these annotations are rejected from the CCDS database.
Types of CCDS QA tests performed prior to acceptance of CCDS candidates
| CCDS QA testa | Test purpose |
|---|---|
| Subject to NMD | Checks for transcripts subject to NMDb, which are unlikely to be coding |
| Quality low | Checks for low coding propensity |
| Has non-consensus splice sites | Checks for non-canonical splice sitesc |
| Predicted pseudogene | Checks for genes that are predicted to be pseudogenes by UCSCd |
| Ortholog not found/not conserved | Checks for genes that are not conserved (UCSC calculation) and/or are not in a HomoloGene clustere |
| Too short | Checks for transcripts or proteins that are unusually short, typically <100 amino acids |
| RefSeq is not an NP_ | Checks if the RefSeq has model (XP_) status; only NCBI matches with NP_ IDs are permitted as CCDS ID accessions |
| CDS start or stop not in alignment | Checks for a start or stop codon in the reference genome sequence |
| Internal stop | Checks for the presence of an internal stop codon in the genomic sequence; possibly a selenocysteine codon or ribosomal frameshift |
| Length mismatch versus genome | Checks if the protein encoded by the reference genome sequence is the same length as the matching annotation sequences |
| NCBI:Ensembl protein length different | Checks if the protein encoded by the NCBI RefSeq is the same length as the EBI/WTSI protein |
| Low percent identity versus genome | Checks for >99% overall identity between the matching annotations and the genomic-encoded protein |
| NCBI:Ensembl low percent identity | Checks for >99% overall identity between the NCBI and EBI/WTSI proteins |
| Accession dead | Checks if an associated RefSeq is no longer valid |
| GeneID changed | Checks if the GeneID has been changed |
| Gene discontinued | Checks if the GeneID is no longer valid |
| Not protein coding | Checks if the GeneID no longer has a protein-coding locus type |
| More than one GeneID represented | Checks for accessions associated with >1 GeneID; allowed only for readthrough genes that encode the same protein as an individual gene |
aAll tests are performed following the annotation comparison step of each CCDS build and are independent of individual annotation group QA tests performed before the annotation comparison.
cSplice donor-acceptor pairs other than GT-AG, GG-AG and AT-AC.
dPredicted retrotransposed genes (9).
eNCBI’s database for the automated detection of homologs (http://www.ncbi.nlm.nih.gov/homologene/).
Some QA tests look for possible contraindications within the coding sequence and its annotated structure that may include identifying issues with the genome sequence. Other types of QA tests assess the quality of the annotation match between the NCBI and Ensembl genome annotation data sets, assess the coding potential for the annotated CDS, assess the possibility that the annotated CDS is more likely a pseudogene and reconcile the annotation data set with current information, especially with regard to genes that have been withdrawn or changed to a non-coding type. Basic integrity checks on the protein products represented ensures that the proteins represented with a CCDS ID have consistent matching in their reading frames and sequences, not merely in genome annotation coordinates. Some sequence differences are expected and tolerated due to a major difference between the NCBI and EBI/WTSI annotation strategies. NCBI reference genome annotation is generated by aligning known RefSeq transcript records to the genome, which is then supplemented by calculating new annotations based on primary sequence data in the INSDC database. In contrast, the manual and computational annotations in the Ensembl data set are direct annotations on the reference genome. Since the NCBI known RefSeq transcript and protein set is generated using transcript data (supplemented with some publication support and personal communications), these records may have some sequence differences, typically small polymorphisms, compared with the reference genome sequence. The majority of RefSeq proteins included in the CCDS data set are identical to the translation derived from the genome sequence, with a total of 781 CCDS IDs (1.6% of the data set) representing curatorial decisions to retain sequence differences in associated RefSeq proteins based on abundance of support evidence, conservation and publication data. The CCDS database, therefore, has some tolerance for minor differences between the NCBI and EBI/WTSI annotations (though not affecting the start, stop or splice site locations), with these differences being due to sequence polymorphisms between the reference genome sequence and the transcript data or publication support the RefSeq is based on.
Review process and curation
An important part of maintaining the integrity of the CCDS database is the constant review of the data, and the ability to make necessary changes to the represented CCDS IDs, which may involve either update or withdrawal of the CCDS ID. Unlike most individual databases, where usually a unilateral curatorial decision is made on a given representation as per the policies of the particular database, the CCDS database is unique in that the review process must be carried out by multiple collaborators, and agreement must be reached before any changes are made. This multi-collaborator involvement in turn contributes to data accuracy and quality and it underscores the ‘gold standard’ definition of the CCDS data set.
Achieving consensus among multiple collaborators presents a challenge, and thus it is necessary to have a good collaborator coordination system that includes a work process flow and forums for analysis and discussion. The CCDS database, therefore, operates an internal website that serves multiple purposes including curator communication, collaborator voting, providing special reports and tracking the status of CCDS representations. For example, the internal website reports CCDS IDs that have been flagged for review by any group for a potential annotation update or withdrawal, tracks CCDS IDs that were recently updated or withdrawn and need an explanatory public note and provides a variety of reports ranging from QA analysis during a CCDS build to reports of all CDS annotations that do not yet have a CCDS ID (Prospective CCDS).
When a collaborating CCDS group member identifies a CCDS ID that may need review, a voting process is employed to decide on the final outcome (Figure 1). Voting is tracked on the internal CCDS website and is carried out by the manual curation groups, RefSeq and HAVANA, as well as by the UCSC group that acts as an independent voice. A consensus agreement is needed to proceed with an annotation update that will alter the CDS structure at a start or stop codon or a splice donor or acceptor site or to withdraw a CCDS ID. In the event of conflicting votes, which may occur due to different curation policies or alternative interpretation of data, further discussion of associated data and/or publications takes place until an agreement can be reached on the CCDS representation. This is supported by an e-mail issue tracking system (Atlassian JIRA) that facilitates tracking a discussion over time, uploading images or files to support a position or retrieving the history of a discussion for a similar case. The same system is used to support enquiries submitted by public users of the CCDS resource via the provided Contact page (http://www.ncbi.nlm.nih.gov/projects/CCDS/UserRequest/UserRequest.cgi). The process of discussing conflicts results in one or more groups modifying their vote in the CCDS internal curation system to reflect the final consensus agreement to update or withdraw a CCDS ID. Each manual annotation group then applies the update independently (i.e. NCBI updates a RefSeq transcript and HAVANA manually updates genome annotation). These updates are integrated into the genome annotation processes at NCBI and Ensembl, resulting annotation is compared and the CCDS analysis process confirms the update and increments the version number of the CCDS ID. If agreement cannot be reached for an update due to either ambiguous support data or different requirements for support evidence by the RefSeq and HAVANA groups, then the default is to withdraw the CCDS ID. In this case, annotation may continue to be provided by one or both annotation groups; however, the annotation is not considered sufficiently supported to be retained in the CCDS database.
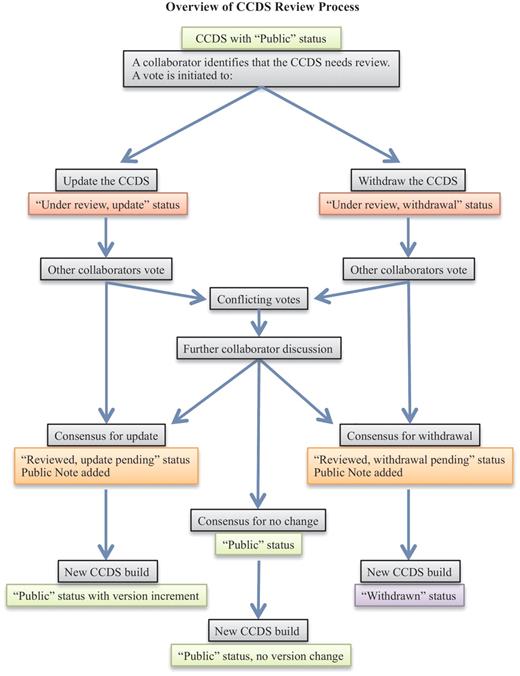
The flowchart outlines the CCDS review process (light gray boxes). CCDS IDs undergo status changes during and following the review process, as indicated by the colored boxes, where light green indicates ‘Public’ status, red indicates an ongoing review that has not yet reached consensus, orange indicates a pending update or withdrawal that has reached consensus, and purple indicates ‘Withdrawn’ status.
Why are curation guidelines necessary?
During manual curation sprints of the CCDS data set, we noticed that some types of manual annotation updates were conflicting between the collaborating groups at a higher frequency, resulting in time-consuming discussions in order to reach agreement. Upon review of the annotation guidelines that were already established by the RefSeq and HAVANA curation groups, it became apparent that these conflicts were often due to either contradictory curation guidelines, or incomplete guidelines. Therefore, it was desirable to establish a common set of curation guidelines to optimize consistency and to minimize the need for further discussions. Although the CCDS collaboration members share the common goal of achieving consistent annotation for protein-coding genes, several factors make this a challenge. First, each member of the CCDS collaboration also employs curation guidelines that were independently developed to address the full spectrum of annotation needs by those groups (broader than the scope of the CCDS collaboration). Second, the collaboration involves multiple annotators working at very different locations from each other. CCDS curation guidelines were therefore established after careful discussion including review of the current literature and are available on the CCDS website. The established guidelines are oriented toward addressing those annotation details that resulted in a higher frequency of conflicting annotation or that consistently required lengthy discussion to reach agreement. These guidelines are used for ongoing curation between CCDS builds and curators refer to specific sections when flagging a CCDS ID for an update or when discussing a more complex case with conflicting opinions. The curation policies established for the CCDS data set have been integrated into the RefSeq and HAVANA annotation guidelines and thus, new annotations provided by both groups are more likely to be concordant and result in addition of a CCDS ID. It is important to note that these standards address specific problem areas, are not a comprehensive set of annotation guidelines, and do not restrict the annotation policies of any collaborating group.
Currently, we have established guidelines for: (i) nonsense-mediated mRNA decay (NMD) candidates, (ii) inhibitory upstream open reading frames, (iii) annotation of translation start sites where there is more than one possible start codon and (iv) management of protein-coding readthrough transcripts. The CCDS curation guidelines are based on known biological principles, experimental results reported in the literature and literature-based guidelines related to current understanding of what ‘typically’ occurs in vivo at the transcript and protein production levels. In many cases, the only definitive solution to a question regarding validity of a distinct protein isoform inferred from aligned transcripts (or an inference regarding the correct translation start site) is experimental data that shows production of specific protein isoforms. In the absence of experimental evidence, curators make an educated inference based on our established guidelines as to the most likely correct CDS representation while acknowledging in a public note that there may be other interpretations.
Curation challenges and guidelines
NMD
NMD is a eukaryotic surveillance pathway that destroys abnormal transcripts containing a premature termination (or stop) codon (PTC) that encode a truncated protein (for reviews, see 10–12). If translated, the resulting aberrant protein may cause disease. The NMD process has a large body of associated research (see reviews), but less is known about the features that the cellular machinery uses to decide whether a transcript should enter the NMD pathway. Different models have been suggested to explain NMD, including the exon junction complex (EJC) model and the ‘faux 3′–UTR’ model (13, 14), but none of these models are entirely satisfactory for explaining when NMD occurs (15, 16). It has been reported that NMD is widespread and ∼45% of human genes have one isoform that is targeted by NMD (7, 8), and for many of these, the pathway is acting to regulate gene expression.
Consequently, NMD is a major consideration for CCDS protein representations, and therefore, we have established curation guidelines to address this issue. A conservative approach is taken for assessing the potential of transcripts to be NMD candidates and potentially produce non-functional proteins. Using the EJC model, if the stop codon is >50 nt upstream of the last exon–exon junction, then the transcript is assumed to be an NMD candidate. Any CDS annotation based on such a transcript is excluded from the CCDS data set except in the following circumstances: (i) If all transcripts at the locus are NMD candidates but the locus is known to be protein coding, then one of these transcripts will be represented in the CCDS data set and (ii) If there is experimental evidence demonstrating that a functional protein is produced from such a transcript.
Historically, NMD candidate transcripts were represented in the CCDS data set as they were annotated as protein coding by both RefSeq and HAVANA. However, this policy was later revised to exclude them from the CCDS data set unless there is evidence for the protein, as indicated above, and proteins associated with NMD transcripts were flagged for review to withdraw them from the CCDS data set. For example, CCDS8237.1, which represented the human KLHL35 gene, was annotated based on the mRNA AK091109.1, which retains an intron introducing a PTC, so it was identified as an NMD candidate after curation guidelines were established (Figure 2). Given the lack of experimental evidence for the protein predicted on this transcript, this CCDS ID was withdrawn. In contrast, CCDS53542.1 representing the human DUSP13 gene is an example of an NMD exception that was retained in the data set because there is publication support (17) for protein production from the CDS.
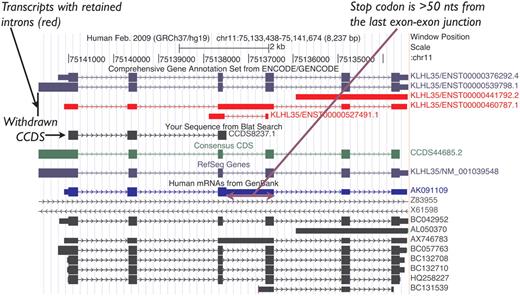
UCSC Genome Browser view of the human KLHL35 (kelch-like 35) gene. CCDS8237.1 was based on AK091109.1 (mRNA track, blue). This CCDS ID has now been withdrawn because a retained intron introduces a premature termination codon, rendering the transcript an NMD candidate. CCDS44685.2 representing the completely processed full-length variant remains valid for this gene.
Consequently, additions to the CCDS data set of this type do not occur by chance because there are established criteria for when a CDS annotation associated with an NMD transcript may be included in the CCDS data set. The RefSeq group also revised its annotation policy such that proteins are only represented for NMD transcripts according to the CCDS guidelines, and all other NMD transcripts are represented as non-coding (with the putative ORF annotated with a misc_feature, see NR_003256.2). The HAVANA project has different goals and annotates these transcripts with a CDS and tags them as NMD candidates for researchers who need them for designing experiments, particularly those in the proteomics field.
Upstream open reading frames
Another consideration for the CCDS data set is the presence of upstream open reading frames (uORFs) that reside upstream of the main or primary ORF (pORF) (reviewed in 18, 19). The scanning model for translation states that the small (40S) ribosomal subunit scans the mRNA starting from the 5′-end and then initiates from the first translation start codon (20), thereby making it possible that a uORF could be translated first, which could then subject the transcript to NMD. In mammals, it is thought that as many as 25% of the genes possess uORFs (21), which may encode bioactive peptides, but it is known that many uORF-containing transcripts can still produce the protein product from the downstream pORF. For example, the human CD1C transcript (RefSeq NM_001765.2, VEGA OTTHUMT00000046351 and Ensembl ENST00000368170 associated with CCDS1175.1) contains four uORFs encoding peptides ranging from 3 to 29 amino acids, one of which has a strong Kozak signal, yet translation of the longer 333 amino acids downstream ORF occurs to produce a functional protein (22). This may be explained by other studies showing that short ORFs of 18–20 codons tend to be resistant to NMD, with 35 amino acids being the approximate size limit for uORFs that escape NMD due to re-initiation of ribosomes at a downstream translation start codon (20, 23, 24).
It is thought that uORFs likely play a role in translational regulation and tend to reduce, but not abolish, translation and that longer uORFs that have a strong Kozak context are more inhibitory (18–20, 25). As with NMD, it would facilitate annotation if there were experimental data available for assessing the impact of each uORF, but currently there are a limited number of genes for which this type of published data is available. A recent genome-wide study showed that upstream ORFs inhibit translation but do not completely abolish it (25). However, the data set in this study was not large enough to provide enough statistical power to study the effect of combinations of features on gene expression and so effects of single features were analyzed in turn, e.g. uORFs with strong Kozak signals were analyzed independently of longer length uORFs, whereas CCDS curators need to consider the combined effects of these features and also the combined effects of multiple uORFs. Additional genome-wide data could therefore facilitate annotation of transcripts with uORFs. Although such data are lacking, we still need to have a joint CCDS policy to address cases where transcripts have uORFs that all groups can agree upon. Currently, the annotation guidelines document includes policies regarding annotation of coding regions based on transcripts containing uORFs which have a strong Kozak signal and are either ≥ 35 amino acids or overlap the pORF. These standards were generated following a review of the literature early on in our collaboration and include consideration of the uORF length, Kozak sequence strength and whether the uORF overlaps the pORF. In light of recent publications, this is an area that is currently under discussion and we anticipate revising the CCDS guidelines in the future.
Multiple in-frame translation start sites
As mentioned above, the scanning model states that the ribosome will initiate translation from the first start codon with a favorable context that it encounters when scanning from the 5′-end of an mRNA sequence (20, 26, 27) which provides the premise for assuming that translation usually starts from the first AUG, e.g. preproinsulin, CCDS7729.1. Translation initiation is influenced by several factors including the length of the 5′ leader sequence (26), uORFs, hairpin secondary structure near the translation initiation site (20, 28, 29) and the sequence context around the translation initiation site. A favorable start codon context is defined based on the Kozak signal for which there is an optimal sequence (gccGCCACCAUGG for vertebrates) (30), although this can vary and a G or A at position −3 and a G at +4 makes the strongest contribution to the context (where the ‘A’ of the AUG is +1) (20, 27). The presence of a U at the +5 position is also thought to negate the effect of G at the +4 position (20, 31). Therefore, the CCDS collaborators define the sequence [A/G]NNaugG[not U] as a strong Kozak signal, with any other sequence being considered a weak Kozak signal.
Exceptions to the rule of translation from the first AUG involve three mechanisms—ribosome re-initiation, leaky scanning and the use of upstream non-AUG start codons (20, 27). Short ORF length may allow reinitiation at a downstream ORF; distance between the upstream and downstream ORFs is important since the ribosome needs time to gain another Met-tRNAi-eIF-2 that is necessary for recognition of an AUG codon (27). A second exception occurs if the AUG is not in a strong Kozak context which may permit leaky scanning (20) by the ribosome, which bypasses this AUG to start translation from a downstream start codon. Potentially, multiple different proteins could be produced from one mRNA and this has been experimentally confirmed for some transcripts (20, 32).
The CCDS guidelines stipulate that the longest ORF should be annotated unless there is compelling evidence that indicates translation initiation from an internal AUG is more likely. The CCDS database represents one translation start site per CCDS ID with the goal to represent the more likely translation initiation site. Thus, if curators think it is possible that additional start codons may be used for translation, this is indicated in a CCDS public note (e.g. CCDS28818.2 representing the mouse Vegfa gene). The current CCDS guidelines are based on principles of the scanning model for translation as discussed above, and they also include considerations for experimental evidence, community standards for start codon annotation, conservation of the start codon and the presence of protein domains or localization signals.
CCDS4929.1 representing the human CRISP3 (cysteine-rich secretory protein 3) gene is an example of how the AUG guidelines were applied (Figure 3). This CCDS ID was originally based on the mRNA X95240.1, and the 5′-most AUG in that transcript was selected as the start codon. However, compared with other available transcript data, this transcript is 5′-partial and its first exon does not extend far enough to include an upstream AUG found in other transcripts. The collaborators, therefore, voted to update this CCDS ID to version 2 to extend the 5′-end of the CDS to the upstream start codon, supported by the mRNA AK292786.1 and increasing the encoded protein length by 13 amino acids. The CRISP3 gene product is known to be secreted (33). The protein predicted from both start sites has a predicted signal peptide (SignalPv4.0). The upstream AUG has a weak Kozak sequence and appears to be primate-specific, whereas the downstream AUG has a stronger Kozak sequence and it is conserved among more mammals. Since there is some conservation of the upstream AUG and the signal peptide for the longer protein is a reasonable size, it is reasonable to annotate the upstream AUG as the start of the CDS and therefore the update was approved. Nonetheless, since the upstream AUG has a weak Kozak signal, it is possible that ribosomes may initiate translation from the downstream start codon at some frequency due to leaky scanning, and both start codons could be used in vivo.
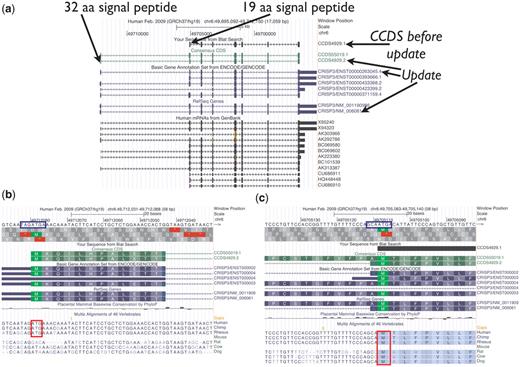
UCSC Genome Browser view of CCDS4929.1, which was updated to version 2, representing a variant of the human CRISP3 (cysteine-rich secretory protein 3) gene. The CDS was extended at the 5′-end. (a) Both the longer protein (258 amino acids) encoded by the update and the shorter protein (245 amino acids) have predicted signal peptides (SignalPv4.0) of 32 amino acids and 19 amino acids, respectively. (b and c) Base-level view. The upstream AUG start codon (b) has the weaker Kozak context (blue box) and is only conserved among primates (red box), whereas the downstream AUG (c) is conserved among more mammals (46-way alignment and conservation track).
Readthrough transcripts
The CCDS collaboration has established guidelines for the treatment of a special class of transcripts, known as readthrough transcripts. Readthrough transcripts arise when transcription initiates in one gene, continues past the normal transcription termination signals for that gene, and terminates within or at the end of a downstream gene on the same strand. Readthrough transcripts may span two or more genes in the same genomic neighborhood. Splicing generates a mature transcript that includes exons from each gene, and may include novel exons from the intergenic region. Such transcripts may encode a fusion protein derived from exons of each gene, or the coding sequence may be in-frame with one gene and have frameshifts with respect to the other gene(s), or the readthrough transcript may possibly be non-coding due to NMD. The biological function of readthrough transcripts and/or the encoded products is not yet understood. While the definition of ‘readthrough’ has been described elsewhere (34), the CCDS collaboration definition of readthrough is very specific in that the individual partner genes must be distinct, and the readthrough transcripts must share ≥1 exon (or ≥2 splice sites except in the case of a shared terminal exon) with each of the distinct shorter loci. Unlike the broader definition of ‘conjoined’ genes described by Prakash et al. (34), the CCDS collaboration readthrough definition does not include cases where the genes are otherwise considered to be co-transcribed (e.g. human HOXC4, HOXC5 and HOXC6) (35), bicistronic (e.g. human CERS1 and GDF1) (36), overlapping each other but not sharing splice sites (e.g. the 3′ exon of the mouse Mon1b gene overlaps the 5′ exon of the Syce1l gene) or genes that have nested arrangements relative to each other (e.g. human and mouse protocadherin gene clusters) (37).
The presence of two distinct genes and readthrough transcripts present some annotation challenges, especially with regard to which gene the readthrough transcripts should be associated with. In consultation with the HUGO Gene Nomenclature Committee (HGNC), the CCDS collaborators have recently agreed, in most circumstances, to represent the readthrough transcript as a separate locus. Several protein-coding readthrough transcripts are represented in the CCDS data set, with each readthrough event having more than one line of independent support to exclude sporadic artifacts. Curation is currently ongoing and has mostly been focused on human genes thus far; it is expected that more readthrough transcripts will be included in the CCDS data set following future builds. An example is CCDS56237.1 representing the ARPC4-TTLL3 gene, which encodes a fusion protein that shares identity with the products of both individual genes.
Discussion
CCDS curation guidelines were established to address specific annotation conflicts that were observed at a higher frequency. These guidelines were guided by experimental data with default options established to define ‘best practice’ approaches when experimental data is not readily available. Establishment of CCDS curation guidelines has helped to make the CCDS curation process more efficient by reducing the number of conflicting votes and time spent in discussion to reach a consensus agreement. In addition, integration of these curation policies into the RefSeq and HAVANA guidelines has resulted in increased consistency for manually annotated CDS regions, with a corresponding increase in the number of proteins tracked with a CCDS ID, and a corresponding reduction in the number of new annotations that end up in the Prospective CCDS report. CCDS curation guidelines are fluid due to the increasing biological research into the issues affecting the ability to accurately represent the structure of genes mapped to the reference genomes, as well as addition of new data that can be used as evidence. Therefore, as biological understanding of translation initiation, NMD and uORFs increases, the curation policies will be reviewed and updated. In the future, genome-wide data sets may help more accurately determine what occurs, in vivo, for each transcript rather than applying generalized rules. Proteomics data could help confirm when alternate in-frame translation start sites are used, or the translation of uORFs.
A major limitation of the CCDS data set is that not all protein-coding loci or coding splice variants are currently represented in the CCDS data set. Although we have established joint CCDS annotation guidelines, they address specific issues as indicated above, and other annotation differences remain. The lack of a CCDS ID for a given gene or CDS could be due to differences regarding the project goals for the RefSeq and HAVANA groups, support evidence requirements, alternate determinations with regard to the protein-coding nature of a transcript or simply due to the fact that one or both groups has not yet have annotated the gene or a particular splice variant. The CCDS genome annotation analysis process identifies proteins annotated by any member of the collaboration for which annotation is not consistent (and thus it will not gain a CCDS ID). These are tracked as Prospective CCDS cases in the internal website with a mechanism for curators to flag annotations that can readily be added to the CCDS data set based on an annotation addition or update by RefSeq or HAVANA. Thus, the project work flow includes periodic focus by curators to proactively address protein-coding genes that lack CCDS IDs; this ongoing curation is facilitated by the established CCDS curation guidelines. Manual monitoring of the Prospective CCDS queue indicates that the use of established CCDS guidelines by RefSeq and HAVANA curation staff is yielding more consistent CDS annotation.
Limitations in supporting data are more difficult to address, such as the lack of sufficient transcript data to define the full-length exon combination. Some protein annotations are intentionally excluded from the CCDS data set due to quality issues with the supporting transcripts or published experimental data, such as retained introns, chimerism or concerns based on a publication description on how a cDNA was cloned, sequenced or assembled, or concerns about the limitations of the experimental approach used. However, for most supporting data, there is no reason to suspect, or else there is insufficient information to determine, that there is a quality concern, and thus the quality of the resulting CCDS representations rely heavily on the quality of the underlying primary data.
Since the CCDS data set represents genomic annotations, quality issues with the reference genome sequence present another challenge. This affects genes that are located in or around gaps in the reference genome assembly, or where the reference genome is misassembled, contains a frameshifting indel, premature stop codon or polymorphic pseudogene and cannot represent the correct protein, e.g. the human NBPF14 gene and polymorphic pseudogene GPR33 (38). CCDS project collaborators report identified problems with the human and mouse reference genome sequence data to the Genome Reference Consortium (GRC, http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/) (39) that investigates and makes a correction, if deemed appropriate. Once the genome problem has been corrected in a new assembly, the gene can then be represented in the CCDS data set, e.g. CCDS45604.1 representing the human FXR2 gene.
It is important for other annotation groups and researchers to understand both the process flow used to generate the CCDS data set and the curation guidelines applied to the manually curated subset of the CCDS data set, as this information guides interpretation and use of the data set. User feedback indicates that the CCDS data set is a valued definition of high confidence coding exons and it is used in large-scale epigenomic studies, production of exon arrays (40), the design of exome capture kits (41) and the design of an in silico set of oligonucleotides (the Human OligoExome) (42). The CCDS data set is also integrated into the GENCODE (http://www.gencodegenes.org) (4) gene annotation project (one of the projects of the ENCODE consortium, http://www.genome.gov/10005107) (43, 44).
Gene annotation continues to be essential for interpretation of the functional elements of the genome, in the study of genome and gene evolution, and for experimental design. Comparative analysis is confounded by application of different annotation standards to different genomes, and thus we feel that the standards being established by the CCDS collaboration should be considered in a wider context. New sequencing technologies have greatly improved the speed while significantly reducing the cost of generating whole-genome sequence data; at the same time new or improved assembly algorithms are more efficiently assembling sequence data into genome assemblies (45). This has resulted in a large expansion in the number of species being sequenced, and this is anticipated to continue to increase as there are a number of projects that aim to sequence the genomes of numerous species such as the Genome 10 K Project (46). The cost of providing manual curation support to annotate these genomes is prohibitive and thus they will be annotated using computational pipelines. As a data set that is more significantly curated and subject to international agreement, we anticipate future use of CCDS data as a quality assurance measure of annotation results. In addition, the curation standards being established for the CCDS project may guide further refinements to computational pipelines to adhere with CCDS project criteria.
Funding
The work done at NCBI was supported by the Intramural Research Program of the National Institutes of Health, National Library of Medicine. Work done at UCSC has been funded with Federal funds from the National Human Genome Research Institute (NHGRI) for the ENCODE project (prime award 5U54 HG004555, under subaward 0244-03 from the Wellcome Trust Sanger Institute). Work done at the Wellcome Trust Sanger Institute has been funded by the Wellcome Trust (grant number WT077198) for HAVANA and by the Wellcome Trust (grant number WT062023) and the National Human Genome Research Institute (grant number 5U54HG00455-04) for Ensembl. Funding for open access charge: the Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest. None declared.
Acknowledgements
We wish to thank the programmers, database and curation staff at Ensembl, NCBI, WTSI and UCSC for their contribution to the CCDS analysis, maintenance and continuing curation efforts. We also thank the UniProt Consortium, HGNC and MGI for many useful discussions that improve protein representation in all data sets. We thank Elspeth Bruford for her input on readthrough transcripts.
References
Author notes
†These authors contributed equally to this work

{kind=link}
{kind=link}
{kind=link}