





Abstract
Manual annotation of genomic data is extremely valuable to produce an accurate reference gene set but is expensive compared with automatic methods and so has been limited to model organisms. Annotation tools that have been developed at the Wellcome Trust Sanger Institute (WTSI, http://www.sanger.ac.uk/.) are being used to fill that gap, as they can be used remotely and so open up viable community annotation collaborations. We introduce the ‘Blessed’ annotator and ‘Gatekeeper’ approach to Community Annotation using the Otterlace/ZMap genome annotation tool. We also describe the strategies adopted for annotation consistency, quality control and viewing of the annotation.
Database URL: http://vega.sanger.ac.uk/index.html
Introduction
High-quality manual annotation of a genome is enormously valuable to aid its interpretation and provide an accurate gene set which serves as a solid foundation for a wide array of further studies, as the value of a genome is only as good as its annotation. Manual annotation can prove to be costly, as it requires a considerable infrastructure, such as a large-scale automated analysis pipeline and specific tools, in order to be viable. The human and vertebrate analysis and annotation (Havana) team at the Wellcome Trust Sanger Institute (WTSI) (1) manually annotate the human, mouse and zebrafish genomes using the Otterlace/ZMap genome annotation tool (2). The manual annotation from the Havana team is released every three months and publicly available from the Vertebrate Genome Annotation Database (VEGA) database (3).
Several genome annotation models were described by Lincoln Stein in 2001 (4):
Museum approach: model organisms with sufficient funding e.g. model organism databases such as flybase (5), wormbase (6).
Party: the jamboree, a short intensive annotation workshop e.g. Drosophila, mouse cDNAs (7,8)
Cottage industry: decentralized effort among several groups of experts e.g. fungal and prokaryotic genomes (9) (10)
Factory: automated genome analysis, used by the genome browsers [Ensembl (11), UCSC (12), NCBI mapViewer (13)].
Despite it being over 10 years since the publication of the Stein paper, manual gene structure annotation is still lacking for many organisms and has been hard to adopt as a community effort because of the limitation of tools available. Where community annotation has been extremely successful is the wikigenes project (14). However, this has been associated with adding descriptive text to attribute functionality to existing gene structures rather than annotation of new gene structures. The main genome browsers have now adopted a mix of factory and museum models, which is employed by NCBI, UCSC, Ensembl and Ensembl Genomes (15). For human annotation, Ensembl and UCSC now display a merged geneset, which is a mix of manual and automated annotation, called the GENCODE geneset (16). This GENCODE annotation will comprise the first pass annotation of the whole of the human genome by the end of 2012. Many genome browsers now also make use of the Distributed Annotation System (DAS) (17) to aid data sharing. This enables browsers to display the most recent data about a region of interest, as DAS makes use of the common reference sequence as a basis to visualize additional annotation. The issue of lag time between browser builds is thus eliminated and data can be accessed and displayed as soon as it is made available to the community.
Community annotation using Otterlace/Zmap
The Otterlace manual annotation system (18) comprises a relational database that stores manual annotation data and supports the graphical interface, Zmap. The Otterlace database schema is based on the Ensembl schema. The annotation data is stored in a MySQL database (19) and forms the backend to the Vega database. ZMap is a stand-alone sequence feature viewer derived from the Acedb FMap display (18). It is written in the C language for high performance and has a command interface so it can be integrated with other annotation software, such as Otterlace. It has a very flexible data model allowing the incorporation of new data sources (e.g. short reads) as they become available.
Funding for manual annotation is limited and therefore we have explored a community annotation approach, which utilizes our annotation software and analysis pipelines. We have used the ‘Blessed Annotator’ and ‘Gatekeeper’ approach within two projects.
Blessed annotator
A variation on the Museum approach. This has been applied to the knockout mouse project (KOMP) (20) and the North American Conditional Mouse Knockout project (NorCOMM) (21). This is part of the International Knockout Mouse Consortium (IKMC) (22) that aims to generate mutants for all of the protein-coding genes in mouse of which WTSI is a member. Since internally, we had developed tools for the analysis of mouse knockout genes for the European Conditional Mouse Mutagenesis Program (EUCOMM) (23), we developed the ‘Blessed Annotator’ approach for KOMP and NorCOMM external annotators. In additional to gene annotation, the mouse projects required the identification of the critical exon; that is the exon in a gene that can be removed to induce Nonsense Mediated Decay (NMD) (24) and so knock-out the expression of that gene. Following on from this, the knock-out construct itself, missing the critical exon, was annotated in order to provide information for the vector constructs that the laboratory partners generate (25).
We conducted extensive training for a small group of annotators from Washington University for KOMP and one annotator at the University of Manitoba for NorCOMM, who were given remote access to our annotation tools so they could continue their work after the initial training period. After a period of close mentoring and quality control (QC), their annotation is considered to be of sufficient quality to be integrated into the mouse gene-build. Both groups have been using our software for 3 years to contribute to their projects.
Gatekeeper
We have also used the ‘Gatekeeper’ approach for multiple species. This is an extension and refinement of the party plus cottage industry approach. We have held several annotation jamborees at WTSI in Hinxton, Xenopus Tropicalis in 2005 (cDNAs), Cow in 2007(WGS) (26) and Pig in 2008 (WGS). These were week-long intensive jamborees to annotate cDNA and genomic sequence with our in-house annotation tools (Otterlace/ZMap), aimed mainly at the Principal Investigators (PI’s) of interested groups. The disadvantage with the jamboree model is that the annotation is a one-off event and the PI’s are usually unable to be available to extend and refine the annotation subsequently, so this is not suitable for a longer-term annotation project. The development and refinement of our annotation tools, which is discussed in the following section, led to their use externally and hence opened up the possibility of external community annotation.
WTSI is involved in the sequencing of the swine genome as part of the swine genome sequencing consortium (27), and is finishing and manually annotating the pig X and Y chromosomes. Our involvement in the annotation in pig and the interest generated by the pig jamborees led to an approach by the Immune Response Annotation Group (IRAG), to annotate ∼1700 genes in pig that are involved in immune response. The genes were chosen by searching for the gene ontology term for immune system process (GO:0002376), core genes involved in host pathogen interplay (28) and gene sets under positive selection in humans (29) within Ensembl (30).
A group of researchers working on resistance to disease and immunity in swine was identified to establish shared and species-specific immune response and to refine the annotation of immunity-related genes. Group training was instigated at WTSI and Iowa State University, with regular follow up meetings by web conferencing tools, such as WebEx and Skype. Groups of researchers were assigned genes of interest and annotated them using Otterlace/ZMap under the instruction and guidance of professional annotation staff.
Software and analysis tools
The Otterlace annotation client runs on a local machine and downloads all of its data from the WTSI web server. The genomic region being annotated is stored in a persistent annotation session directory on the user's computer, which can be recovered following system reboots. Annotation actions require only occasional network access, so the system is tolerant of interruptions to network connectivity.
The genomic sequence is run through an analysis pipeline that consists of homology searches, gene predictions and de novo sequence analysis. The pipeline analysis includes: BLASTX against SwissProt and TrEmbl proteins, BLASTN against ESTs and vertebrate mRNAs, tandem repeat finder, Augustus (31)and Genscan (32) gene predictions. The results are displayed in the ZMap graphical interface (Figure 1B). ZMap is written in the C programming language to give good drawing performance and makes use of threading to load multiple datasets simultaneously resulting in much faster startup times.
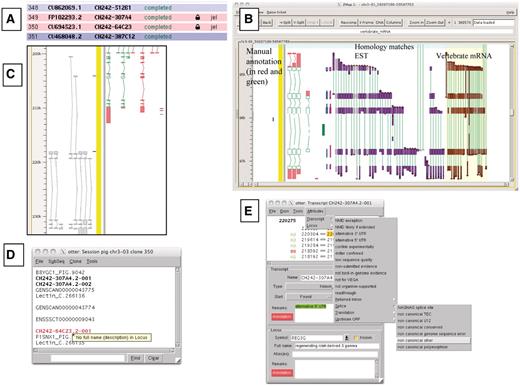
A selection of different views of Otterlace and ZMap. (A) Assembly sequence chooser showing user’s email displayed on locked clones. (B) ZMap view of the results of pipeline analysis, namely EST (in purple) and vertebrate mRNA (in brown) homology matches together with manually annotated transcripts (in red and green). (C) Manual annotation shown as ‘greyed out’, non-editable transcripts where they extend past the genomic region that has been opened. (D) Internal QC displaying a ‘tool tip’. (E) Transcript editing window showing a non-consensus splice site that has been highlighted in red, and a selection of attributes available at the transcript level (green shaded text).
Large-scale data analysis, such as searches of mRNA libraries against the whole genome, are performed on WTSI systems, served by Otter CGI scripts, and presented in ZMap on the client where they can then be used to construct the annotation. Additional sources of evidence, such as BAM files on FTP or web servers anywhere in the world, can be configured on the server and then loaded into ZMap for display. As many of these data sources can be very large ZMap allows the annotator to choose which tracks and how much of each track is loaded.
Access to the Otter system is restricted to authorized users. External annotators register themselves with the WTSI SingleSignOn system, using their email address at their Institute. This takes care of authentication, and access to each species (authorization) is controlled via a configuration file which lists their email address and which is administered by the Otter support staff at WTSI.
Users save annotation back to the master Otter annotation databases. Since it contains a relatively small quantity of valuable data, this database is carefully and frequently backed up. Saving edits to genes does not delete old versions, but writes new versions of genes into the Otter database. It is therefore possible to recover old versions of genes if mistakes are made. The author of any changes to genes and transcripts is recorded, so who has been editing what is tracked. Unchanged transcripts keep their author, but changed transcripts are given the new author, and the author of the parent gene changes to the new author too. The system tracks changes to genes and transcripts via their stable identifiers, and these are shown on the VEGA (33) and Ensembl websites too. These stable identifiers remain attached to each version of genes and transcripts stored in the database, and are independent of any changes to their names.
Locks are used to prevent more than one annotator making changes to the same region of the genome (Figure 1A). Existing genes which are not contained entirely within the region being annotated cannot be edited in the otterlace session and appear ‘greyed out’ (Figure 1C).
Quality control in Otterlace
The Otterlace client performs a number of quality and sanity checks as genes and transcripts are built by the annotator. The names of transcripts with problems are highlighted in red in the session window, and a ‘tool tip’ gives a brief description of the problem when the annotator mouses over the transcript name (Figure 1D). The transcript editing window shows the 2 bp in the intron immediately adjacent to each exon, and colours them green if they match a splice consensus, and red if they do not (Figure 1E). Introns are checked to make sure that they are not too short. When present, the protein translation is checked for internal stop codons and completeness, and the transcript is checked to ensure that it is not subject to NMD (34), or if it is subject to NMD has been correctly flagged. The format of the transcript name is checked to ensure that it conforms to an approved naming convention. Transcripts must have evidence attached (accessions of the nucleotide or protein sequences used to build them), and more than one transcript in the same gene cannot share the same evidence. The locus must have the full name associated with the gene symbol added in the Full name field. A vocabulary of attributes, which can be attached to transcripts or loci is provided to avoid keying errors, and these appear in the transcript window with green shading (Figure 1E).
This integrated QC within Otterlace proved a valuable tool for external annotators as it flags errors as they occur and reduces the need for QC by Havana annotators. For the Blessed annotator model, due to the extended training period there is minimal manual QC over a period of several years for several thousand genes. However, for the Gatekeeper annotator model, the manual QC is much more extensive due to the much shorter training period of the annotators. Thus, this model requires more frequent input by professional annotators but over a shorter timescale compared to the KOMP and NorCOMM projects. The annotators were all trained with reference to the Havana team annotation guidelines (35) which was very important to give an assurance of the quality of the annotation.
The annotation
The annotation for the KOMP and NorCOMM projects took advantage of the customized software features that were already available for the EUCOMM project (25) in particular identifying critical exons and making knock-out constructs. The number of genes targetted for annotation is 5000 for KOMP and 500 for NorCOM, and they are complimentary to the EUCOMM project. This Blessed annotation makes use of the full complement of biotypes that are available within Otterlace, and is integrated into the gene set for mouse that is available from the VEGA website. Gene target for knockouts are identified from Ensembl predictions. Figure 2 gives and example of the importance of manual annotation for this project.
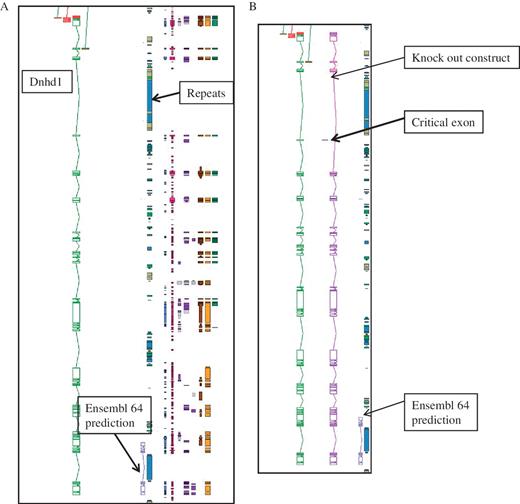
An example of manual annotation in mouse to identify a critical exon. (A) Dnhd1 is a KOMP target gene that is automatically chosen to create a knockout from the Ensembl prediction. A Zmap view of the Dnhd1 gene manually annotated in mouse. The Ensembl 64 prediction is partial, probably due to the highly repetitive nature of the genomic region. (B) A Zmap view of the position of the critical exon identified after completion fo the manual annotation and the associate knockout construct. If the Ensembl prediction had been used as a model to create the knockout a rage proportion of the gene would have been missed. This would have resulted in an unsuccessful knockout construct.
The IRAG project has ∼30 external annotators working through a list of ∼1700 genes. For the pig project a condensed version of the biotypes was used due to the dearth of sequence evidence available for pig and the lower quality of the genome sequence. The reduced numbers of pig mRNA and SwissProt entries that are available and required to make a coding locus biotype Known_CDS, resulted in many more Novel_CDS made from cross-species mRNA evidence. Working with unfinished genomic contigs was a challenge for both the software and the annotators, as for high quality finished genomes, such as human, the annotation is added to finished BAC sequences. For the pig autosomes many BACs consist of several, often unordered, contigs that are not finished to a high quality. Figure 3 shows and example of how manual annotation can assist in assessing the quality of a genome assembly.
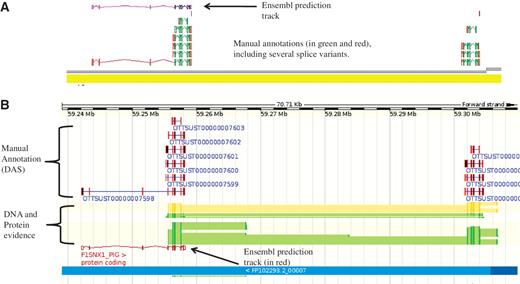
An example of manually annotated genes viewed in ZMap and also displayed as a DAS track in Ensembl. (A) ZMap view of copies of the REG3G gene in pig. The automated Ensembl track predicts one copy of the gene, whilst the manual annotation can resolve two copies in this section of the genome. (B) Ensembl view of the same region displaying the manual annotation DAS track.
In order to find genuine deletions and duplications of pig genes relative to the human genome, a high-quality genome is required. The current pig assembly 9.2, is thought to be missing ∼10% of the genome. The process of gene annotation identifies assembly and sequencing errors, but as full finishing will only be performed on the X chromosome it is unlikely that these errors will be resolved under current plans.
Despite the concerns about the quality of the genome, with reference to high-quality manual annotation, the group has already identified at least 12 genes that show genuine duplication, for example the REG3A gene. Genes that are thought to be absent in the swine genome will be re-assessed when the new genome build is available to ensure that they are not artefactual deletions.
The HUGO Gene Nomenclature Committee (HGNC), (36,37) naming convention for pig genes orthologous to human was used whenever possible and the Havana naming convention for potentially duplicated/similar genes was followed (see guidelines). The KOMP, NorCOMM and IRAG projects are ongoing and the number of de novo genes annotated to date are 1876, 378 and 1276 respectively. The full swine genome is not available in VEGA so in order to view the manual annotation a DAS track for Havana Pig manual annotations is available in Ensembl, called ‘havana_pig’ and can be found from the DAS source http://das.sanger.ac.uk/das/havana_pig. An example of this can be seen in Figure 4.
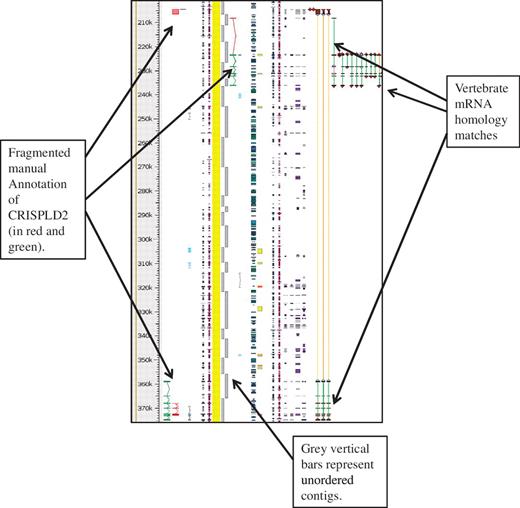
Unordered contigs on pig chromosome 6 viewed in Zmap. The annotation of the CRISPLD2 gene shows clearly how the annotation highlights the fragmented nature of the assembly and aids in identifying the correct contig ordering. The vertebrate mRNA homology matches show the mismatches in the contiguity of the sequence. If sequence is contiguous the connecting lines between the matches are green, but where there is missing or incorrectly ordered sequence the connecting lines are orange.
Discussion
Compared with other community annotation projects, it is apparent that the ‘Blessed Annotator’ and ‘Gatekeeper’ models can give a much wider range of biotypes with a relatively small number of annotators. This comparison is shown in Table 1. The majority of the projects, including Methanosarcina acetivorans, cow and Bee, only annotate Coding genes. The Drosophila project includes non-protein coding RNAs, as does rice. Drosophila also include pseudogenes, but admit that it has very low pseudogene numbers (17 in the whole genome). The rice project gives a five-level Coding gene breakdown, but these are based on automated annotation with a final manual curation step. The annotation using Otterlace/ZMap includes the full biotypes that we have developed in the Havana team. Please see the annotation guidelines for further information (35) These include:
Coding loci: Known_CDS, Novel_CDS, Putative_CDS, NMD.
Non-coding genes: retained_intron, lincRNA, antisense, sense_intronic, sense_overlapping, 3′_overlapping_ncRNA.
Pseudogenes: Processed_pseudogene, transcribed_processed_pseudogene unprocessed_pseudogene, transcribed_unprocessed_pseudogene, unitary_pseudogene, transcribed_unitary_pseudogene, polymorphic_pseudogene.
We also annotate polyA signal and sites to ensure that we have annotated the full-length gene. These detailed biotypes give a much more informative picture of the genome and increases the value of the manual annotation when compared to other annotation methods.
A comparison of a selection of community genome annotation projects
| Organism | Reference | Website | Tool | Number of annotators | Annotation biotypes |
|---|---|---|---|---|---|
| Bee | Elsik et al., 2006 (39) | http://hymenopteragenome.org/beebase/ | Website and Apollo | 177 (61) | Coding |
| Drosophila | Misra et al., 2002 (40) | http://flybase.org/ | Apollo | Unknown | Coding, non-protein coding RNA, pseudogenes (17) |
| Cow | Elsik et al., 2009 (26) | http://genomes.arc.georgetown.edu/drupal/bovine/ | Apollo and Otterlace | 150 | Coding |
| M. acetivorans C2A | Galagan et al., 2002 (9) | broadinstitute.org/ | Calhoun | 30 | Coding |
| Rice | Itoh et al., 2007 (41) | http://rice.plantbiology.msu.edu/ | Automated, then ORFs curated into five categories. | Unknown | Coding, non-protein coding RNA |
| Pig | n/a | http://www.animalgenome.org/pig/ | Otterlace/Zmap | 30 | Full Havana biotypes |
| Mouse (KOMP and NorCOMM) | Skarnes et al., 2011 (25) | http://www.knockoutmouse.org/ | Otterlace/Zmap | 3 | Full Havana biotypes |
The goal of both of our approaches to community annotation is to manually annotate all of the genes required by the projects, with a much depth of detail as possible. Due to the use of the SingleSignOn system for access to the annotation tools, we can track authorship and as mutiple users are prevented from annotating the same region of a genome at the same time there is no duplication of annotation. We routinely transfer annotation over to new genome builds when they are available. For the IRAG project, the annotation will be transferred over to build 10.2 and the annotation reviewed and checked for additional supporting evidence. This is particularly important where genes are partial or thought to be duplicated or deleted with reference to the human genome due to the unfinished nature of the pig genome. This may cause transfer issues where contigs have been re-ordered with relation to the previous genome build and in cases where new sequence has been incorporated into the new build. The use of annotation guidelines is essential to ensure consistency throughout the annotation of all annotation groups, although discussion is valuable when appropriate to aid in their interpretation. The Gatekeeper annotation approach is particularly challenging, as consistent and timely QC is required to address the diverse levels of experience and expertise throughout the group. This method is being successfully adopted by the Bovine Genome Database, which is using the annotation tool Apollo to allow collaborators to annotate new gene structures (38) and a professional curator to validate the data.
It is essential to ensure consistency in gene naming and the IRAG project has provided a good start to establishing this for the pig and thus has highlighted the need for a Swine Genome Nomenclature Committee. This has also demonstrated the added value of manual annotation compared to automated annotation, to give accurate gene structures and gene locations and aid the production of a reference gene set. A possible next step in this community annotation effort could be the annotation of gene families across the genomes of multiple species by utilising the experience of experts in the field. We are also looking at using the information from RNA-seq to confirm and expand alternative transcripts is many different tissue types.
Funding
This work was supported by the Wellcome Trust (grant number WT077198). Funding for open access charge: BBSRC grant number: BB/F02195X/1
Conflict of interest. None declared.
Acknowledgements
We would like to thank Professor Christopher Tuggle and Dr Claire Rogel-Gaillard, the members of the KOMP, NorCOMM and IRAG annotation groups and the Havana annotation team.

{kind=link}
{kind=link}
{kind=link}
{kind=link}