





Abstract
Molecular dynamics simulations can now routinely generate data sets of several hundreds of gigabytes in size. The ability to generate this data has become easier over recent years and the rate of data production is likely to increase rapidly in the near future. One major problem associated with this vast amount of data is how to store it in a way that it can be easily retrieved at a later date. The obvious answer to this problem is a database. However, a key issue in the development and maintenance of such a database is its sustainability, which in turn depends on the ease of the deposition and retrieval process. Encouraging users to care about meta-data is difficult and thus the success of any storage system will ultimately depend on how well used by end-users the system is. In this respect we suggest that even a minimal amount of metadata if stored in a sensible fashion is useful, if only at the level of individual research groups. We discuss here, a simple database system which we call ‘Bookshelf’, that uses python in conjunction with a mysql database to provide an extremely simple system for curating and keeping track of molecular simulation data. It provides a user-friendly, scriptable solution to the common problem amongst biomolecular simulation laboratories; the storage, logging and subsequent retrieval of large numbers of simulations.
Download URL:http://sbcb.bioch.ox.ac.uk/bookshelf/
Introduction
Biomolecular simulation research is capable of generating vast amounts of data. For example, molecular dynamics (MD) simulations generate trajectory files that provide a time-dependent description of the atomic positions while Monte Carlo (MC) simulations generate files that provide an ensemble description of atomic positions. The resulting amount of data from these methods is currently of the order of hundreds of gigabytes and is likely to continue to increase. In recent years there has been much emphasis on the curation and accessibility of scientific data, particularly from research councils [for example, see the BBSRCs data sharing statement issued in 2007 (1)]. Much of the data that is generated is in fact accessible on request directly from research laboratories, but the trend has been to develop databases that provide this information direct to the biochemical community (2,3). Such databases can provide an easy means for curation and storage and also tracking of simulation data.
Previous efforts such as BioSimGrid have concentrated on developing a robust framework for the deposition, analysis and remote retrieval of simulation data (4–6). A particular success of the BioSimGrid project was the ability to perform direct comparative analysis of simulation data (7). One important aspect that became apparent from that project was that the process of deposition has to be user-friendly. In other words, the deposition of the data for a user has to be made as simple as possible. If it is perceived in any way to be cumbersome or difficult, users will simply not make use of such a facility even if they are able to fully appreciate the worth of such a system. This is particularly important for the sustainability of these databases (8) as clearly, a database is only as useful as the data it contains. Ease of use is an often underestimated factor, but is critical because it involves trying to elicit a change in the culture about the way in which simulation data is managed. A similar problem has been discussed in terms of implementing electronic laboratory notebooks (ELNs) (9). An alternative but similar approach was used in the Dynameomics project (10). The Dynameomics project constructed a database built around a large set of simulations with a common theme. Both the BioSimGrid and Dynameomics projects had technical commonalities; both stored all raw data in the database (with consequences for the speed of access as well as the level of detail which can be stored) and both explicitly aimed to perform analyses on the stored data in the database. An even more ambitious project was the Glyco-MGrid project (11) designed for glycobiology simulations, which had the additional aim of allowing users to perform additional simulations and analysis on deposited data. Despite these efforts, there does not exist, at present, any lightweight tools designed to integrate biomolecular simulation data into a workflow with minimal additional effort. Although similar tools were used within the BioSimGrid and Dynameomics projects, they were not released publically. Such tools, which could offer an unambiguous, unalterable link between a publication or presentation and a set of simulations would offer an invaluable tool to principle investigators, and may become more important as simulations are performed in an increasingly high-throughput manner (12,13)
Here, we describe a simple approach, called ‘Bookshelf’, that can be used by individual research laboratories to help keep track of their own simulation data via an easy to use framework for the deposition of local trajectories. The emphasis is on providing a tool for research leaders to track simulation data as it is generated within their group and while it explicitly only aims to aid the storage of large data sets, we suggest that it provides a basic strategy for tracking simulation data in a research laboratory. It differs from the philosophy behind BioSimGrid in that it is not a grid-enabled database and thus does not suffer from the problems associated with middleware that such projects were prone to.
Bookshelf is written in python and allows for the deposition of trajectories from MD simulations generated in house to be curated locally. The users can search the database using keywords and retrieve the files to analyse the trajectories and rerun the simulation or run an extension of the current simulation with modified input parameters. We have the installation process straightforward with minimal dependencies. A further difference between this approach and BioSimGrid, is that Bookshelf does not convert any data formats, thus it does not require updating when individual codes update their own trajectory formats for example. The development of such a database will facilitate the comparison of the results of MD simulation (7,14) and we describe below how this has already been achieved in the context of Bookshelf. We also describe the work-flow of the database and demonstrate the process of deposition and retrieval.
Design philosophy
The underlying philosophy of Bookshelf is that the system should encapsulate the minimal amount of information needed to run that simulation in as simple a fashion as possible. The goal is not to provide world-wide access to all of the simulation data that a research group produces. Rather, it is to supply a simple tool for laboratory heads (and users) to curate and manage their data. The emphasis is on simplicity and ease of use. Most users of molecular simulation codes are used to using a command line interface. Thus if they are able to enter a one-line command to ‘deposit’ their simulation data, this would be preferable to the time-consuming and laborious process of filling in numerous boxes in a web-form for example. Use of a command-line deposition procedure also enables integration with existing scripts that are typically used in a simulation-based laboratory and in turn facilitates the deposition of several simulation sets at the same time. Similarly, retrieval can also be incorporated into such scripts which might facilitate a user’s own record-keeping. For example, integration into a wiki is straightforward. Another aspect of Bookshelf that is a key feature of the design is the ability to allow users to simply take a ‘copy’ of a simulation if they wish to do further analysis or repurpose some aspect of a particular simulation for a new project.
Although a command-line interface facilitates deposition and specific retrieval requests, laboratory heads typically prefer an overview of all or large sections of their simulation data and for this purpose a web interface usually provides the quickest way of assessing the content of the database.
Implementation
The overall architecture is shown in Figure 1. At the data storage level we made the decision to separate the raw data files and the metadata (data ‘about’ the simulation). In essence the actual raw data is copied into a directory with a unique identifier (name). In our case, this directory is located on our hierarchical filesystem (HFS), but in practice this could be anywhere with enough capacity (and where the data is safe). This provides the additional advantage that users have easy and direct access to the raw trajectory data rather than having to formally extract the data via a database query. The actual copy process is handled by a daemon to facilitate protected ownership of the ‘deposited’ data; once the data is written by a user, only a privileged user can remove or change it. The daemon and the associated deposition scripts are written in Python (15). The deposition scripts transfer the metadata and the unique trajectory identifier into a MySql database (16). The metadata can be queried via a python environment or via a web-interface.
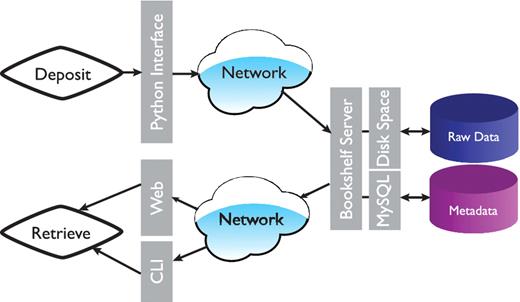
Overall architecture of the Bookshelf system. Command line interface is represented by CLI.
Deposition
The user deposits the data using a simple python script. The script takes the project name, the program used to generate the trajectory and comments about the trajectory as arguments followed by files to be deposited. The project name can be used in any way that the user sees fit; it may describe a methodological study or it may be used to identify a particular protein to which the simulation refers. We have not restricted its use in any way, but it is a requirement to have something that describes the data in some way. There is also the option to provide a doi or pubmed identifier for the published data. The data is stored with a unique id called TrajId. It currently accepts the trajectories generated by different simulation packages like GROMACS (17), CHARMM (18), NAMD (19) and data from the ab initio code, Gaussian (20) but it will be trivial to add support for other data. Depending on the program name supplied, the script expects the user to deposit a program-specific set of essential files that are necessary to reproduce that data. Additional files can also be deposited just by adding them to the end of the command.
For example, the command to run the script for the deposition of a gromacs-based simulation would be:
$ bookshelf.deposition.py -m <project name> -p <program name> -c <‘comments’> -d <doi> -I <pubmed id> <test.top> <test.mdp> <test.xtc> <test.pdb> <optional_file_1>…
Potential of mean force calculations, which typically comprise several windows of simulation data for a particular pathway, can be deposited as one simulation set. The individual windows are assumed to reside within one folder, which can be specified with the deposition command as follows:
$ bookshelf.deposition.py -m <project name> -p <program name> -c <"comments"> -f <yes> <folder name>
Data retrieval
The meta-data can be queried via a simple python based command:
An example text-based retrieval is shown in Figure 2.$ bookshelf.browse.py -s <keyword>
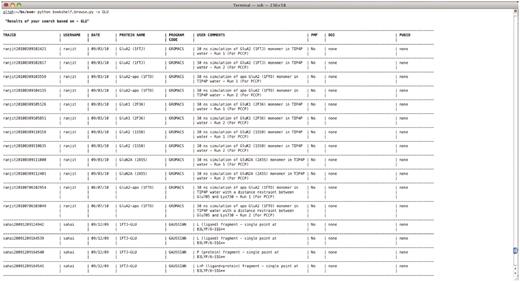
Output from a retrieval command ‘bookshelf.browse.py –s GLU’.
The meta-data in the database can also be accessed through the web interface either by browsing on fields or searching it by using keyword. The database can be browsed by user name, date, program name and protein name. It also gives a summary of the number of trajectories in Bookshelf based on user and the program code. Example snapshots of the web interface are shown in Figure 3. As the raw simulation data is stored as a flat file on a locally accessible disk, it can readily be retrieved with the usual UNIX cp command.
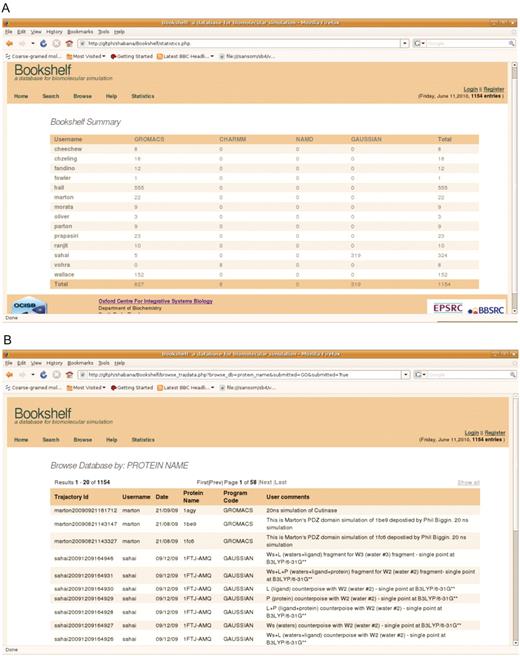
(A) Summary page describing depositions per user. (B) The web-interface displaying a metadata query by protein name.
Support for other formats
We recognized that for this to be useful in a general way, we had to make the process of adding support for any simulation package as simple as possible. The requirements for each package are stored as XML files and a new package can be added quite easily by running the following command:
where ‘program name’ is the name of software package one wants to add; ext1, ext2, etc. are the extensions for the mandatory files that need to be submitted for the deposition. The script adds the package to the existing list that the deposition and retrieval tools can handle.$ add.program.py –p <program name> <.ext1> <.ext2> <.ext3> <.ext4>
Example deployment
Most of the development was done in the Oxford laboratory, but it was clear that the tools would be immediately beneficial for work performed in Southampton and thus this provided an ideal opportunity to test deployment ‘in the wild’. It was successfully deployed (see Figure 4 for output from that installation) and indeed is now being used to help keep track of simulation data generated as part of a collaboration between that laboratory in Southampton and another academic laboratory in the UK. Both laboratories are interested in understanding the role of dynamics in a family of proteins known as ‘Autotransporters’, which form part of the Type V secretory pathway in bacteria. The proteins use a C-terminal ‘translocator’ β-barrel domain to secrete a ‘passenger’ domain in a way that is, so far, poorly characterized. The research groups are hoping to use simulation data in a comparative manner in order to provide insight into the mechanism of action for this class of protein. It is also being used to track previous published simulation data (21). As an additional by-product of that deployment, the tools are now Mac OS-X compatible.

Example output from the installation at Southampton.
Conclusion
We have developed an application for the easy deposition and the retrieval of the trajectories generated by MD simulations. Deposition and retrieval of the data is via a python-based script system. A web-interface is also developed to have easy access to the metadata. Hence, Bookshelf provides a simple means for research groups to manage their data more efficiently and provide an additional layer of accountability allowing researchers to link a given set of data to specific publications. As the metadata is stored in a MySql database it should be possible to integrate it quite easily into the ELNs or wiki technologies that are emerging or indeed run it on increasingly popular decentralized ‘cloud’ services such as Amazon’s E3 and SC2 services. The biggest challenge appears to be changing the culture of data management at the individual level. Such a phenomenon has been observed for ELNs and indeed the situation is particularly problematic in academic environments. A recent report suggested that only 4% of academic laboratories use an ELN (22). However, the advantages are clear in that it encourages standard operating procedures and the dissemination of best practice within a laboratory. The application approach here is quite simple and it would be straightforward to adapt it to other types of biochemical data.
The python code and installation instructions are available for download from:
Funding
Oxford Centre for Integrative Systems Biology (BBSRC); Wellcome Trust (to P.C.B.).
Acknowledgements
P.C.B. and S.K. are both RCUK Fellows.

{kind=link}
{kind=link}
{kind=link}
{kind=link}