





Abstract
Data generation, driven by rapid advances in genomic technologies, is fast outpacing our analysis capabilities. Faced with this flood of data, more hardware and software resources are added to accommodate data sets whose structure has not specifically been designed for analysis. This leads to unnecessarily lengthy processing times and excessive data handling and storage costs. Current efforts to address this have centered on developing new indexing schemas and analysis algorithms, whereas the root of the problem lies in the format of the data itself. We have developed a new data structure for storing and analyzing genotype and phenotype data. By leveraging data normalization techniques, database management system capabilities and the use of a novel multi-table, multidimensional database structure we have eliminated the following: (i) unnecessarily large data set size due to high levels of redundancy, (ii) sequential access to these data sets and (iii) common bottlenecks in analysis times. The resulting novel data structure horizontally divides the data to circumvent traditional problems associated with the use of databases for very large genomic data sets. The resulting data set required 86% less disk space and performed analytical calculations 6248 times faster compared to a standard approach without any loss of information.
Database URL:http://castor.pharmacogenomics.ca
Introduction
Since the release of a working draft of the human genome project, there has been a proliferation of technologies to perform large-scale genotyping. The research possibilities provided by genome-wide analysis have created a data deluge reminiscent of Moore’s Law (1). In a single year, one massively parallel sequencing machine can produce nearly nine times the amount of data currently housed in the US Library of Congress (2–4).
Currently, lengthy analysis times required for the vast quantities of genotype data generated make interactive analysis impractical (3). Sequential access, such as retrieving data from flat files, e.g. PLINK input files (5), has the limitation that all prior data must be processed in order to access datum at the end of the file, and this process must be repeated for each variation of the original analysis. Furthermore, they are memory-bound. Although PLINK provides a solution for many users there are no fixed limits to the size of the data file (5). Larger data sets will require an ever increasing amount of RAM. For example, a sample set of 20 000 individuals and 1-million SNPs would require ∼8–16 GB of RAM (http://pngu.mgh.harvard.edu/∼purcell/plink/faq.shtml#faq5).
Alternately, the use of databases has been hampered due to challenges in data loading time and performance. The development of new techniques and tools is therefore necessary, as historical solutions have been rendered impractical due to the extreme volume of data generated (2–4).
Efforts at reworking this analysis process have focused on three main areas: data structures, data indexing and data analysis. Initially, the improvement of data structures began with a logical model of genomic and phenotypic data using object-oriented structures (2,6), relational databases (2), or mark-up languages (7), which add a lot of model description metadata. These structures are more suited to providing data context and long-term storage than high-speed analysis (8), although some allow basic analytical querying (9). Recent data indexing efforts seek to improve pattern or sequence search performance. While these have shown a significant performance increase for specific targeted tasks, they have the drawback of increasing the data set size by up to 10 times (10–12). Hardware-based solutions such as cloud computing, peer-to-peer networks, and other distributed computational concepts are now used to prolong the useful lifespan of software by increasing processing power. Other solutions circumvent the problem of large data sets altogether at the cost of losing content (13). Current data warehousing and data sharing methodologies are making progress but fall short of providing a solution for rapid analysis (14).
There are three obvious areas for improvement: (i) reduce data set sizes without any loss of information (also reducing long-term data storage costs), (ii) eliminate the need for sequential access and (iii) organize data to allow for rapid analysis. Our solution attempts to address all these areas of concern. Using established computer science principles we have developed the comprehensive analysis and storage (CASTOR) methodology, a normalized, multi-table and multidimensional database structure for storing and analyzing genotype and phenotype data.
Methods
Data normalization
Data normalization restructuring techniques reduce redundancy and increase the flexibility of a poorly structured data set without loss of information (15). These techniques are frequently used to make a data structure suitable for implementation in a relational database management system (RDBMS). Common genomic data sets such as Illumina’s Genomestudio output files, certain PLINK input files and the Gencode GTF format are all examples of data structures that, despite being produced by, or for, automated analysis contain a significant amount of redundant data and therefore violate the principles of normalization.
In a typical Illumina Genomestudio results file 63% of the output file is composed of unnecessarily redundant data. Although only a single instance of each datum is required to communicate the necessary information, fields such as sample identifier, the name of the single nucleotide polymorphism (SNP) in question, and SNP position are needlessly repeated for each row contained in the sequential file (Table 1). Since both SNP name and SNP position are associated with the SNP in question and not the sample, their inclusion on each row violates the second normal form. Because of this, data which should take up a total of 17-million characters (9 character SNP name + 8 character SNP location = 17 characters × 1 000 000 SNPs), or 0.009% of the final data set, instead takes up 119-billion characters (17 characters × 1 000 000 SNPs × 7000 samples), or 63% of the final data set.
Genomic data structure with a large amount of duplicate data
| Sample identifier | SNP | SNP value | SNP position |
|---|---|---|---|
| Sample 1 | rs3094315 | CC | 742 429 |
| Sample 1 | rs41480945 | CC | 21 227 772 |
| Sample 1 | rs4040617 | CG | 95 952 929 |
| Sample 2 | rs3094315 | TT | 742 429 |
| Sample 2 | rs41480945 | AT | 21 227 772 |
| Sample 2 | rs4040617 | CC | 95 952 929 |
This data set is a result of a combination of two different data structures: SNP information (SNP and SNP positions) and sample information (sample identifier) in order to accommodate one piece of datum that depends on both (SNP value).
In order to address this redundancy, we have separated the data set into two individual but related tables. A SNP reference table, containing a list of all SNPs used in the study and their associated position, and a genotype results table containing sample information and all genotypic results.
The SNP reference table uses SNP name as the primary key and related fields as non-prime attributes. This results in one row of information for each SNP present in the study. The genotype results table contains a single row for each sample in the study, with each column representing the results of an individual SNP. This format is similar to the PLINK PED file format (5), which also has one sample per row, using columns to represent the SNPs. This approach leads to a large number of columns. A study involving 1 000 000 SNPs would result in a data set with 1 000 001 columns (one column for sample id, and one column for each SNP). This is impractical as a sequential file, and impossible to implement as a database structure, as a table with these dimensions is not supported by any current database management system (DBMS).
Multi-table
While a DBMS cannot accommodate an unlimited number of columns per table, most can accommodate a nearly unlimited number of tables per database. The number of tables is limited by the capacity of the underlying filesystem or, in the case of Microsoft’s SQL Server, by the number of database objects permitted (over 2 billion). It is this property that we exploit to accommodate our 1 million SNP wide structure, horizontally dividing the single, large, genotype results table of 1 million SNPS into 2000 tables, each with 501 columns (500 SNPs and a sample identifier as primary key). This new structure is currently supported by all major DBMSs.
Each column in the genotype results table is denoted generically (snp1, snp2, etc.) and is included in the SNP reference table allowing rapid identification of the specific SNP. In doing so, uploading a new data set would require no structure changes (such as renaming each column).
Multidimensional encoding
Multidimensional databases are optimized for rapid and ad hoc computer-aided analysis or online analytical processing (OLAP) (16) by encoding all alphanumeric data as numeric data, and isolating descriptive data from the data required for the analysis. Using this methodology we have divided the information into dimension tables and fact tables. Dimension tables contain descriptive data including all the original alphanumeric descriptors and the code that replaces them in the fact tables. The fact tables contain only numeric data and are used to conduct the bulk of the analysis. Each possible combination of two alleles is encoded numerically into 10 values (Table 2).
Genotype dimension table (see genotypes_dim in Figure 1)
| Code | Genotype | Allele_a | Allele_c | Allele_g | Allele_t |
|---|---|---|---|---|---|
| 1 | AA | 2 | 0 | 0 | 0 |
| 2 | CC | 0 | 2 | 0 | 0 |
| 3 | GG | 0 | 0 | 2 | 0 |
| 4 | TT | 0 | 0 | 0 | 2 |
| 5 | AC | 1 | 1 | 0 | 0 |
| 6 | AG | 1 | 0 | 1 | 0 |
| 7 | AT | 1 | 0 | 0 | 1 |
| 8 | CG | 0 | 1 | 1 | 0 |
| 9 | CT | 0 | 1 | 0 | 1 |
| 10 | GT | 0 | 0 | 1 | 1 |
This encoding results in a smaller, faster, and more flexible data set, which is more suitable for analysis. While the structure and content change, none of the information contained in the initial data set is lost.
Phenotypes are similarly encoded. Phenotype data already in numeric format remains unchanged; however, an entry is made in the phenotype_dim table (Table 3) to ensure that the context of the phenotype is not lost. Each alphanumeric phenotype is assigned an integer code in the phenotypes_discrete_dim table and a parent entry is added to the phenotypes_dim table (Tables 3 and 4). Numeric codes from phenotypes_discrete_dim are used to populate the phenotype fact tables. Using this methodology, almost all alphanumeric values are converted to numeric values, making these tables suitable for automated analysis. Note that free-form text entries however, cannot be encoded in this way and therefore should be avoided whenever possible if automated analysis is the goal.
Phenotype dimension table (see phenotypes_dim, Figure 1)
| Id | Name | Discrete | Description | Column |
|---|---|---|---|---|
| 1 | Medication dosing (units) | 0 | Medication dose per day in units | ptype1 |
| 2 | Pain severity | 0 | Severity of patient pain | ptype2 |
| 3 | Smoking status | 1 | Never, former, current | ptype3 |
Discrete phenotype dimension table (see phenotypes_discrete_dim, Figure 1)
| Code | Phenotypes_dim_id | Label |
|---|---|---|
| 1 | 3 | Never smoked |
| 2 | 3 | Former smoker |
| 3 | 3 | Current smoker |
Test platforms
The test platforms for all tests were Dell 2× Quad Core Xeon E54102 × 6 MB cache, 2.33 GHz, 1333 MHz FSB, PE2900, with 16 GB 667 MHz Dual Ranked DIMMS and 8 ×300 GB 15K RPM SCSI 3 Gbps mounted in RAID 1+0 for 1.2 mirrored terabytes of disk space. The operating system was RedHat Enterprise Linux 5, and the MySQL Community Server 5.0.67 compiled for RHEL5 (MyISAM) or Oracle 11G were used as the DBMS.
Tests were conducted using both DBMSs, but only Oracle 11G was able to manage the 7 billion rows contained in the original data set, and therefore was used for all load, statistical and data return comparisons on the original data set.
Two computer hosts were used. The first host handles only the dimension tables and the software client responsible for issuing the database queries and the collection of results. The second host, configured to maximize the performance of the DBMS responsible for manipulating the fact tables, performs the analysis.
Evaluation
In the absence of a sufficiently large publicly available data set, a very large data set composed of 7000 subjects, each with 7000 phenotypes (both quantitative and dichotomous) and 1 000 000 bi-allelic genotypes for a total of 7 049 000 000 data points was randomly generated and used to evaluate the performance of our novel database structure. A test suite was written in Perl (17), which created the database structure, disabled indices before the data set was loaded, loaded the data set and then re-enabled the indices. Load time was defined as the sum of the time required to perform these operations.
Our test suite then measured the impact of the new schema using the same computer hardware and operating system, for a direct comparison. To avoid comparing the speed of a DBMS versus a sequential file, which would require the evaluation of a great number of hardware and operating system variables, the original data set was also loaded into the DBMS for evaluation (see Supplemental Data for more detail).
The database management system and multidimensional nature of the data were kept constant for both the original and CASTOR data sets to measure only the efficiencies of the novel database structure. A variety of statistical and common GWAS analyses were performed on the data sets (mean, square root, minimum, standard deviation, variance, allele count with a phenotypic filter) (Table 5). In addition, we tested how rapidly data could be located and retrieved from the databases.
Query return times of common statistical analyses based on a single table query (genotype or phenotype)
| Query | CASTOR (s) | Original (s) |
|---|---|---|
| Query (gtypes) avg(int) | 0.347017 | 871.454132 |
| Query (gtypes) sqrt(int) | 0.096701 | 0.050104 |
| Query (gtypes) min(int) | 0.319485 | 716.520514 |
| Query (ptypes) stddev(int) | 0.014837 | 1341.081771 |
| Query (ptypes) avg(float) | 0.010675 | 1417.641397 |
| Query (ptypes) sqrt(float) | 0.003062 | 12.227921 |
| Query (ptypes) min(float) | 0.009296 | 0.014992 |
| Query (ptypes) stddev(float) | 0.013895 | 3.202807 |
| Query (ptypes) var_pop(float) | 0.010164 | 16.41966 |
| Query (gtypes) count(int) where int is 1 | 0.325984 | 16.669058 |
| Query (gtypes) count(int) where int is 3 | 0.358017 | 641.80022 |
| Query (gtypes) count(int) where int is 4 and patient_id = 1234 | 0.027244 | 668.470442 |
All queries performed on the Oracle 11G DBMS.
Results
Our CASTOR approach converts the sequential file into a normalized and indexed, direct-access database (Figure 1). Combining all normalization techniques the data set was reduced from 98.4 to 13.8 GB, a decrease in disk space usage of 86%, without loss of information (Table 6). Removing redundant SNP information alone reclaimed over 50 GB of space.
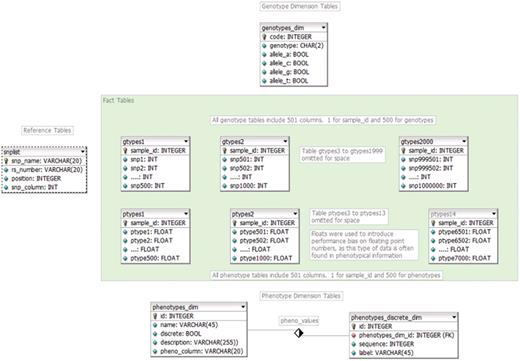
CASTOR data diagram.
Performance comparison results
| Original | CASTOR | |
|---|---|---|
| Size of genotype data | 97 GB | 6.8 MB × 2000 tables = 13.3 GB |
| Size of phenotype data | 1.4 GB | 34 MB × 14 tables = 476 MB |
| Total data set size | 98.4 GB | 13.776 GB |
| Oracle 11G: load time (min) | 493.5 (8.23 h) | 90.3 (1.51 h) |
| Oracle 11G: Total time to run all performance tests (min) | 937.2 (15.62 h) | 0.15 (9.1 s) |
| Oracle 11G: Total time to perform evaluation (min) | 1430.7 (23.85 h) | 90.18 (1.50 h) |
Once loaded, the data can be reused and reanalyzed without the need to repeat either the conversion or the data load. The significant decrease in load time (90.3 min versus 8.23 h for the original data set) is primarily a result of data set size reduction due to normalization, and the corresponding reduction in index size due to the horizontal segmentation of this data set. The smaller indices are easily loaded into available memory when needed, removing the need to use slower hard disk based virtual memory space often required by larger indices.
As each column in the CASTOR data structure represents a SNP, the database metadata itself is responsible for SNP indexing thus obviating the need to separately index the SNPs for rapid data access, as has been the focus of earlier efforts (10–12).
The genotype table from the non-optimized original database (single table) structure had a row count of 7 billion (1 million genotypes for 7000 samples) and the phenotype table had 49-million rows (7000 phenotypes for 7000 samples). With an alphanumeric index (such as the combination of SNP name and sample id), the index alone would take up 111 GB of memory (9 character SNP name + 8 character sample id = 17 characters × 7 billion records). Our CASTOR database has significantly fewer rows (7000 per table, one row per sample), but has 2000 genotype tables and 14 phenotype tables, dividing the data set into smaller, fragmented indices. Using a single row per sample, the index on each CASTOR table is 55 Kb (7000 samples × 8 character sample id) allowing for very rapid load times. The total size of the CASTOR indices (across all 2000 genotype tables) is 107 megabytes, but since the indices are fragmented across many tables, only those indices needed to fulfill a specific query are loaded at any given time. Table 6 illustrates the benefits of the CASTOR approach.
Since DBMSs are optimized for column-oriented calculations, using each column as a list of all genotypes for a particular SNP (SNPs across samples), optimizes the data set for GWAS-type analyses while still supporting row-based calculations across SNPs when necessary.
The final result is a CASTOR data set (containing all of the original information) that is very wide, comprising over 2000 tables and 1-million columns for genotypes alone, but quite short, with only a single row per sample in each table (Figure 1). The resulting multi-table data schema’s time required to conduct the performance analysis was reduced by 99.9% by moving from a single table to a multi-table data structure (15.62 hours compared to 9.1 seconds) (Table 6).
Discussion
Applying both well-known and novel data transformation and data architecture techniques, we have arrived at a simple and elegant solution that achieves a significant data set size reduction and a dramatic increase in processing speed. As data is loaded into the database the data is normalized to remove duplications, then encoded into numerical data and subsequently divided into the novel multidimensional multi-table structure specifically designed for large genetic data set analysis. Converting the original data set into a multidimensional data set has many advantages, such as enabling the use of OLAP (16) and increasing the speed of the data set (Table 5) by eliminating slower alphanumeric data from the analysis tasks. An additional benefit is the further reduction of the size of the data set while preserving all of the information contained in the original.
A multi-dimensional encoding scheme can furthermore be used to encode more than just the initial data. For example, the genotypes dimension table (Table 2) not only encodes the 10 possibilities of genotype pairs, it also easily separates homozygous pairs (code ≤4) from heterozygous pairs (code ≥5). Counting alleles, a basic calculation in a GWAS, can be accommodated with the following structured query language (SQL) query:
select sum(genotypes_dim.allele_a) from genotypes_dim, gtypes1 where gtypes1.snp2 = genotypes_dim.code and genotypes_dim.allele_a > 0
Where genotypes_dim is the database table that holds the information for each genotype; allele_a is a count of A alleles in a particular genotype; gtypes1 is the table containing the genotypes for the first 500 SNPs; snp2 is the field containing the genotype code for snp2.
If adopted, this approach would offload basic statistical manipulations to the database, provide a platform for automated initial quality control and analysis, and result in savings in disk storage, data archiving and transfer time. Our CASTOR approach, if adopted for biological data sets, would provide a much more reasonable starting point that could enable analytical solutions on laptop computers or other non-specialized hardware, while still benefitting from the performance improvements available to cloud computing and other hardware-based solutions. The CASTOR approach will help meet the demand for high-speed analysis by providing a solid foundation to handle ever-increasing amounts of genetic data. Our data set can scale to several million samples and nearly an unlimited amount of SNPs with nothing more than a linear impact on performance.
Aside from the stated performance benefits, CASTOR also has a potential impact on storage costs associated with this data. Based on published estimates, the average long-term storage cost currently is $25/month/GB (18), including all overheads. Prior to any normalization, the original data set composed of 7000 samples with 1-million SNPs and 7000 phenotypes would cost $29 520 per year in total storage costs. Based on the same published estimates, the same information in CASTOR format would cost $4140 per year.
The next step is to incorporate the CASTOR approach into commonly used software packages such as PLINK. The CASTOR approach, as it is DBMS-based, readily accommodates multi-processing and multi-core processor architecture. This should significantly reduce the time required to perform GWAS or similar analyses, allow for the development of new algorithms, as well as extend the lifespan of current software tools by eliminating hardware bottlenecks.
Funding
Génome Québec. Funding for open access charge: Génome Québec.
Conflict of interest. None declared.
Acknowledgements
Thanks to Christopher Beck, Amr Al Mallah and Madjid Hihi for their advice and encouragement; Paul Guelpa for his graphical skills and feedback; and Andrea Smith for comments and suggestions.
References
Author notes
Present address: Sharon Marsh and Tibor van Rooij, Faculty of Pharmacy and Pharmaceutical Sciences, 3126 Dentistry/Pharmacy Centre, University of Alberta, Edmonton, Alberta, T6G 2N8 Canada.

{kind=link}