





Abstract
GeneCards (www.genecards.org) is a comprehensive, authoritative compendium of annotative information about human genes, widely used for nearly 15 years. Its gene-centric content is automatically mined and integrated from over 80 digital sources, resulting in a web-based deep-linked card for each of >73 000 human gene entries, encompassing the following categories: protein coding, pseudogene, RNA gene, genetic locus, cluster and uncategorized. We now introduce GeneCards Version 3, featuring a speedy and sophisticated search engine and a revamped, technologically enabling infrastructure, catering to the expanding needs of biomedical researchers. A key focus is on gene-set analyses, which leverage GeneCards’ unique wealth of combinatorial annotations. These include the GeneALaCart batch query facility, which tabulates user-selected annotations for multiple genes and GeneDecks, which identifies similar genes with shared annotations, and finds set-shared annotations by descriptor enrichment analysis. Such set-centric features address a host of applications, including microarray data analysis, cross-database annotation mapping and gene-disorder associations for drug targeting. We highlight the new Version 3 database architecture, its multi-faceted search engine, and its semi-automated quality assurance system. Data enhancements include an expanded visualization of gene expression patterns in normal and cancer tissues, an integrated alternative splicing pattern display, and augmented multi-source SNPs and pathways sections. GeneCards now provides direct links to gene-related research reagents such as antibodies, recombinant proteins, DNA clones and inhibitory RNAs and features gene-related drugs and compounds lists. We also portray the GeneCards Inferred Functionality Score annotation landscape tool for scoring a gene’s functional information status. Finally, we delineate examples of applications and collaborations that have benefited from the GeneCards suite.
Database URL:www.genecards.org
Introduction
With the recent accumulation of data from worldwide genome projects, the individual scientist faces the time consuming and laborious task of sifting through the expanding labyrinth of gene information. This can be partly alleviated by the use of sophisticated integrated and searchable databases. For many years, GeneCards® (www.genecards.org) (1–3) has provided such a remedy, with carefully selected, comprehensive information about human genes, mined and integrated from over 80 data sources. By bringing together gene information from large public sources such as HGNC (4), NCBI (5), ENSEMBL (6) and UniProtKB (7), as well as many other smaller resources (8), GeneCards has provided concise genome, proteome, transcriptome, disease and function data on all known and predicted human genes. It has successfully overcome barriers of data format heterogeneity using standard nomenclature, especially HUGO nomenclature committee approved gene symbols (4). The information is organized in a ‘card’ format for each gene, in distinct functional sections and including a variety of features such as textual summaries and links to other genome-wide and specialized databases. GeneCards has evolved significantly since initially described (1,9,10), and its progress is documented in a number of past publications (2,3,11–15). In this article, we introduce the new GeneCards Version 3 (V3) and describe its features in detail. We place special emphasis on the novel set-centric capabilities (beyond and in conjunction with the new GeneCards search engine), which address a variety of applications, including microarray data analysis, cross-database annotation mapping and gene-disorder associations for drug targeting.
Readers who are new to GeneCards might want to read the Applications section below first, familiarize themselves with previous articles (1–3), and then read the rest of this article, possibly skipping the ‘Methods’ section.
GeneCards version 3
The new home page
The new GeneCards V3 home page, shown in Figure 1, hosts the new search facility, provides links to a sample gene and its various sections on the card via labeled oval buttons, and enables one to view a variety of differently categorized and annotated genes, from pre-defined links as well as by interacting with a random-gene generator, customizable by category and/or GeneCards Inferred Functionality Score (GIFtS). The GIFtS algorithm (11) uses the wealth of GeneCards annotations to produce annotation scores aimed at predicting the degree of a gene’s functionality. Since the degree of known functionality is correlated with the amount of research done on a particular gene or its product, these annotation scores are presented as inferred functionality measures. The extended GIFtS tool, linked to from the home page, facilitates browsing the human genome by searching for the annotation level of a specified gene, retrieving a list of genes within a specified range of GIFtS values, obtaining random genes with a specific GIFtS value, and experimenting with the GIFtS weighting algorithm for a variety of annotation categories. The left hand side of the home page retains the logos and links to the GeneCards suites sites—GeneDecks, GeneALaCart, GeneLoc, GeneNote, GeneAnnot and GeneTide.
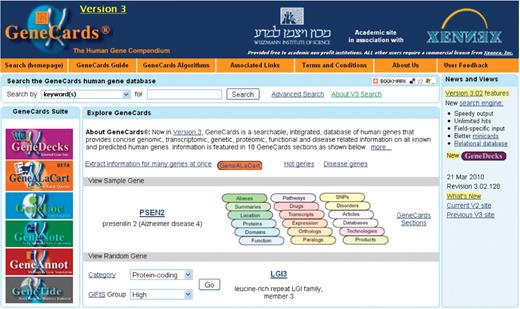
GeneCards Version 3 home page, including search, sample gene, logos and links to the other suite sites, and category/GIFtS-based random gene generator.
The new search engine
The new version 3 search engine is extremely fast, and is capable of matching complex field-specific queries of the entire database in milliseconds. For example, a search for a very common keyword like ‘cancer’ returns ∼8000 results in 3 ms. In contrast, V2 could not handle such a query, or even a more focused one such as ‘melanoma’ (‘too many results to be efficiently displayed’); a considerably more restricted search in V2 such as ‘schizophrenia’ yielded ∼1100 results and took 80 s. Efficient V3 performance is achieved by breaking the search process into distinct phases, and also by returning results in limited pages of data. The two primary stages of each search are: (i) to first quickly identify the list of genes that have information matching the search term, and (ii) upon demand, discover the detailed relevant context and annotation details of those hits, and highlight them in ‘minicards’ (Figure 2). The ‘Methods’ section details the design of the new search engine.

GeneCards Version 3 search results, including detailed minicard expansions which highlight where in the ‘card’ the hits occur.
The upgraded GeneCards webcard
The ‘card’ presented for a GeneCards gene has grown considerably since last described in the literature (1–3). The colored ovals in Figure 1 depict the various sections (aliases, summaries, location, proteins, domains, function, pathways, drugs, transcripts, expression, orthologs, paralogs, SNPs, disorders, articles, databases, technologies and products) wherein relevant data sources are excerpted from and/or deep-linked to in each GeneCards gene. We now highlight some of the updated sections’ interesting content and algorithms.
Header
The header at the top of each GeneCard provides the gene’s symbol, category, GIFtS (11) and GeneCards identifier (GCid) (12). Gene categories of protein coding, pseudogene, RNA gene, cluster, genetic locus and uncategorized, are color coded, with the gene’s symbol painted accordingly. The background color of the header’s box is indicative of which database the symbol is from [HGNC (4), Entrez Gene (5) or Ensembl (6)]. The header also contains a short description of the gene, and spells out whether or not the gene symbol is HGNC approved. GCids, provided by the GeneLoc algorithm (12) are unique, informative and trackable. The id begins with GC, which is followed by the chromosome number, ‘P’ or ‘M’ for orientation (Plus or Minus strand), and approximate start coordinate in kilobases if relevant. When a location is not possible to determine, a sequential number is used in that part of the GCid. If more than one gene falls on the same kilobase, the closest free identifier is chosen. For example, GC09P139152, the GCid for GRIN1, is on chromosome 9 on the plus strand, starting at ∼139 152 kb. While GCids may change from version to version to reflect the reality of new genome builds, once a GCid is assigned to a gene, the association remains (as a ‘previous GCid’), and it cannot be reassigned to another gene. Figure 3 shows examples of a few GeneCards genes, comprising a variety of categories and source databases, along with their GCids and GIFtS, as well as statistics (available at the bottom of the GeneCards home page) about the number of genes per category, with examples from each one.
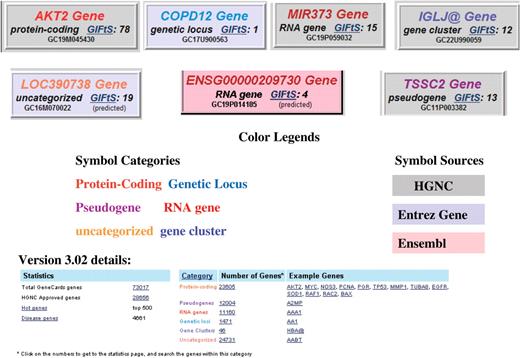
Assorted GeneCards genes, of different color-coded categories, source databases, GC identifiers and GIFtS, with associated statistics and examples.
Proteins
This section provides annotated information of the proteins encoded by GeneCards genes according to UniProtKB (7), and/or Ensembl (6), the capability to view phosphorylation sites using Phosphosite (16), reference sequences (RefSeq) according to NCBI (17), cellular component ontologies visualized by the Gene Ontology (GO) Consortium (18), and links for ordering antibodies, recombinant proteins and assays from numerous sources. Direct links to 3D visualization of PDB structures are provided by the OCA browser (19) and Proteopedia (20).
Gene function
This section provides annotated information about gene function from UniProtKB (7) and Genatlas (21), animal model information from MGI (22), RNAi, primers and clones products from vendors, as well as molecular function ontologies visualized by the GO Consortium (18). While the ‘Orthologies’ section in GeneCards presents a table of orthologs from HomoloGene, SGD, euGenes and MGI, showing symbol, locus, description, similarity to human and NCBI accessions, the Gene Function section presents animal model information from MGI, including mutant phenotypes for mouse orthologs, and a popup table with information on their alleles including: (i) allele name—the official symbol for the allele with link to the MGI record, (ii) the MGI identifier of the allele, (iii) type of allele by mode of origin and (iv) phenotypic details for all genotypes that include at least one of the alleles. We believe that the use of mouse phenotypes to depict human gene function is unique to GeneCards.
Pathways and interactions
This section provides links to—according to information extracted from Invitrogen (23), Millipore (24), Sigma-Aldrich (25), Applied Biosystems (26), Cell Signaling Technology (CST) (27) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (28). For each of the pathway sources, one can also view other genes that participate in these pathways via the GeneDecks (15) Partner Hunter link. Next, a link to the relevant SABiosciences (29) interacting genes and proteins network is provided. Finally, interacting proteins are presented in a table which merges protein–protein interaction data from UniprotKB (7), EBI-IntAct (30), String (31) and MINT (32) including links to the GeneCards gene and the UniProtKB (7) and/or Ensembl (6) protein entry for the interacting protein, as well as detailed annotation about the interactions, including all supporting experiments and/or confidence scores about predicted interactions. Finally, biological process ontologies visualized by the GO Consortium (GO)(18) are presented.
Drugs and compounds
This section provides relationships between GeneCards genes and chemical compounds and drugs, in a similar manner as described below for disorders for the NovoSeek (33) and PharmGKB (34) sources. It also juxtaposes compound names, actions and chemical abstract numbers provided by commercial sources, with links for ordering products. We have found that the richness of the integrated descriptor set of drugs and compounds has enabled unique results for our GeneDecks partner hunting and set distillation subsystems (described below) as compared with comparable gene-set analysis tools (15).
Transcripts
Figure 4 presents the ‘Alternative Splicing’ subsection, with alternative splicing information and isoforms from ASD (35). Exons with alternative splice sites in different isoforms were broken into Exonic Units (coined ExUns). The letters indicate the order of the ExUns in the exon. The symbol between ExUns indicates an intron, while ‘·’ indicates the junction of two ExUns. Mouseovers on the dark blue squares show the ExUn’s genomic coordinates, while mouseovers on the light blue squares show its transcript coordinates. When showing ASD’s splice variants, GeneCards subtracts the 3000-bp flank that ASD adds to the transcript coordinates. The section also displays multiple transcripts from RefSeq and Ensembl.
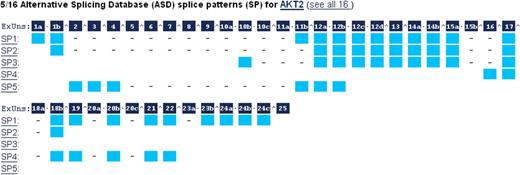
Alternative splicing diagram in the Transcripts section. Exons with alternative splice sites in different isoforms are broken into Exonic Units (ExUns). The symbol between ExUns indicates an intron, while ‘·’ indicates the junction of two ExUns.
Gene expression
Figure 5 depicts the enhanced GeneCards experimental tissue vectors. The same set of non-fetal normal and cancer human tissues are also analyzed and presented in upgraded electronic northern and Serial Analysis of Gene Expression (SAGE) graphs (3). For the experimental data, duplicate measurements were obtained for 12 normal human tissues (out of 28 tissues shown) hybridized against Affymetrix GeneChips HG-U95A-E (GeneNote data) and for 22 normal human tissues hybridized against HG-U133A (GNF data (36)). The intensity values (shown on the y-axis) were first averaged between duplicates; then, probe-set values were averaged per gene, global median-normalized and scaled to have the same median of about 70 (half-way between GeneNote and GNF medians). GNF HG-U133A expression data for 18 NCI60 cancer cell lines was processed and added to the display (a single measurement taken; normalized according to the GNF normal data). The correspondence between cell lines and tissues is given in a table (37). Note that the diamonds along the x-axis of each graph hint that the tissue (cell line) expression values are available for a given gene, while empty ‘diamonds‘ denote that either there is no such tissue for a specific microarray, SAGE or electronic northern platform or that the current gene has no matching probe sets (or tags/ESTs for SAGE/electronic northern). If there is a filled diamond along the x-axis but no data shown in the graph, it indicates that after threshholding and normalization there is no meaningful expression data for that tissue. Normalized intensities are drawn on a root scale (3), which is an intermediate between log and linear scales. The Affymetrix MAS5 algorithm was used for array processing.
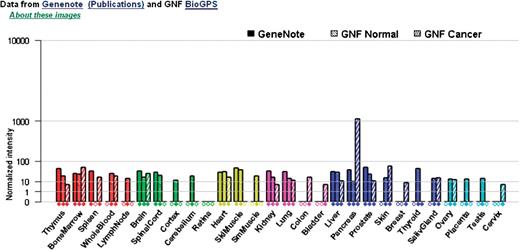
Enhanced experimental tissue vectors, now including our GeneNote data integrated with normal and cancer data from the Genomics Institute of the Novartis Research Foundation (GNF).
SNPs
This section (Figure 6) integrates SNPs/variants data from the NCBI SNP Database (38), Ensembl (6) and Pupasuite (39), and adds descriptions from UniProtKB (7) and linkage disequilibrium images from HapMap (40). Filtering is done to include only those that are not artifacts, not connected to gene duplication, not withdrawn by NCBI, fully specified, without ambiguous locations or low map quality, and having single Entrez Gene and contig ids. The order of a gene’s displayed SNPs can be determined by the user. By default, SNPs are sorted first (shown in the select box as 1st) by validation status (‘validated’ before ‘non-validated’), then, within these groups, by ordered location type (first ‘coding non-synonymous’, then ‘coding synonymous’, followed by ‘coding’, ‘splice site’, ‘mRNA-UTR’, ‘intron’, ‘locus’, ‘reference’ and/or ‘exception’), as the secondary (2nd) nested criterion, and finally, by the number of validations (up to 4). The user can change this default sort order and define up to three hierarchical sorting priorities from fields available as select boxes above the relevant columns on the section’s button line as follows: rs-numbers (sorted in ascending order), validation status, position on the chromosome (ascending order), location type, allele frequencies (existing info before non-existing), population types (alphabetical order) and total sample size (largest to smallest). Each displayed line includes genomic, expression and allele frequency data sections. Only the summary is shown for the expression and allele frequency sections, with a link to the detailed information (via a magnifying glass icon).
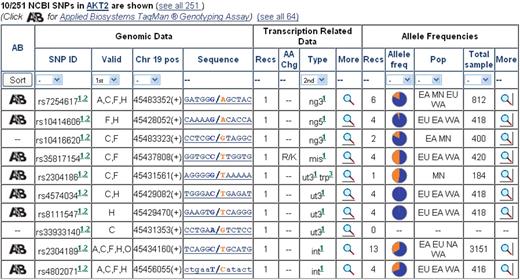
Snapshot of the SNPs section, highlighting the variety of annotation fields, availability of popups for more detailed information, sort options.
Disorders and mutations
This section contains disorders and mutations in which GeneCards genes are involved, according to a variety of sources including OMIM (41), UniProtKB (7), NovoSeek (33), PharmGKB (34), Genatlas (21), GeneTests (42), HGMD (43) and GAD (44). Included are two disease relationships table: The Novoseek table includes: (i) Disease—the name of the disease related to this GeneCards gene. (ii) Score—the Novoseek score of the relevance of the disease to this gene, based on their literature text-mining algorithms. (iii) Articles—the number of articles in which both the gene’s symbol or description and the disease appear. (iv) PubMed IDs for Articles with Shared Sentences (# sentences) - PubMed IDs of articles in which both the gene symbol and the disease appear in the same sentence, sorted by the number of sentences (shown in parentheses in the column) in which they both appear. Similarly, the PharmGKB table includes: (i) disease—the name of the disease related to this GeneCards gene, (ii) the PharmGKB description of the relationship between the gene and the disease, one of the following types: CO, clinical outcome; PD, pharmacodynamics and drug response; PK, pharmacokinetics; FA, molecular and cellular functional assays; or GN, genotype and (iii) PubMed IDs for articles supporting these relationships since both the gene symbol and the disease are discussed.
Research reagents
Distributed among the various sections, and highlighted in the Services section, GeneCards provides directly targeted deep links to cutting edge research reagents and tools such as antibodies, recombinant proteins, primers, clones, expression assays and RNAi reagents. Adding this functionality has proven to be a win–win strategy for the product providers, for the GeneCards project and for researchers; the links have been received very favorably by our users.
GeneCards suite
Gene set analyses via GeneDecks
GeneDecks exploits GeneCards’ unique wealth of combinatorial annotations to identify similar (partner) genes, and to perform quantitative descriptor enrichment analyses for identifying set-shared annotations. Some of these capabilities have been implemented on the GeneCards web site, some as independent research studies, and others are in preparation for upcoming releases:
Annotation combinatorics
Given a particular GeneCards gene, one can ‘GeneDecks‘ it with respect to a selected combinatorial annotation in order to obtain a set of similar genes. The resulting GeneDecks summary table ranks the degree of similarity between the identified genes and the probe gene, taking into consideration all shared combinations of annotations. Thus, if a particular probe gene has N annotations in a given category (e.g. is involved in N pathways or has N domains), GeneDecks separately depicts sets of genes associated with any combination of M of the annotations, 1 ≤ M ≤ N, in descending order of M, thereby highlighting a rank order of similarity. This feature is available on the V2 web-site and within V3’s Partner Hunter’s algorithm.
Annotation unification
GeneCards is replete with annotations from different sources, often with heterogeneous naming conventions. For example, there are several independent systems for pathway definition, each having its own nomenclature and relevant sets of genes, and currently addressed separately when GeneDecksing from the V2 webcard. Figure 7 describes a pilot study that uses similarity measures within GeneDecks gene sets to see if and how the nomenclatures and entities of KEGG (28), Invitrogen (23) and CST (27) independent systems of pathway annotations could be unified. We found that some of the pathways (named identically or differently) had considerable overlap in their gene-set composition, but none were complete, that some of the pathways were closely related, and that a few inter and intra-source subsets could be identified. Annotation unification of this sort, based on the similarity in GeneCards gene-content space detection algorithms, could be expanded to include other [e.g. our Millipore (24) and Sigma-Aldrich (25)] pathways, and perhaps also be generalized to include other attributes such as chemical compounds, phenotypes and/or orthologies.
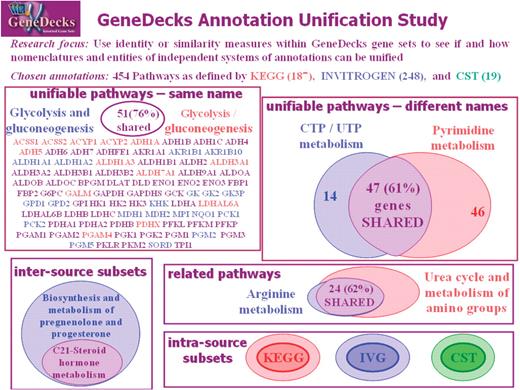
GeneDecks pathway annotation unification study, aimed at matching differently-named pathways based on overlaps in associated gene-set space.
Partner hunting
GeneDecks’s Partner Hunter (15) seeks similar genes based on combinatorial similarity of weighted attributes. It currently addresses gene sets using information for pathways, protein domains, GO terms, mouse phenotype, mRNA expression patterns, disorders, drug relationships and (sequence-based) paralogs.
Set distillation
Set distiller ranks attributes by their degree of sharing within a given gene set. Like Partner Hunter, it enables sophisticated investigation of a variety of gene sets, of diverse origins, for discovering and elucidating relevant biological patterns, thus enhancing systematic genomics and systems biology scrutiny. Both subsystems have been used in the study of synthetic lethality [see ‘Applications, advantages and future directions’ section below and (15)].
Batch queries via GeneALaCart
GeneALaCart provides batch query support, whereby the user submits a gene list (e.g. from a microarray experiment or from search query results), along with the desired GeneCards annotation fields, and receives tabulated output which can be visually examined or serve as input to automated scripts for more sophisticated analyses. Figure 8 is an example of a GeneALaCart results file (45), showing a few of the ∼50 available annotation categories for a small set of genes. The ‘Applications, advantages and future directions’ section below details a variety of examples of research facilitated by GeneALaCart.
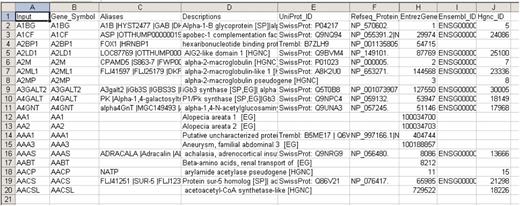
Small sample of GeneALaCart output to a batch query. The data can be examined in Excel or serve as input to application-specific computer analysis programs.
Methods
GeneCards system architecture
Figure 9 depicts the architecture of the offline and online components that comprise the GeneCards system. This is described in some detail in the subsections below.
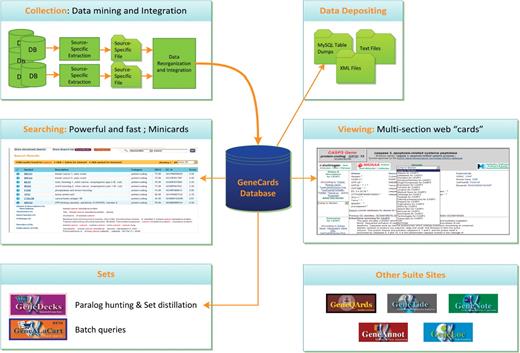
GeneCards architecture and data flow, including offline data collection/ integration and quality assurance processes, relational database, sophisticated search engine and set-centric GeneALaCart and GeneDecks subsystems.
Data collection and integration
The data collection process is a pipeline that starts with defining the full set of GeneCards genes, obtained from three primary sources as follows. First, the complete current snapshot of HGNC-approved symbols (4) is used as the core gene list. Next, human Entrez Gene (5) entries that are different from the HGNC genes are added. Finally, human Ensembl (6) records are matched against the emerging gene list via our GeneLoc’s exon-based unification algorithm (12); those that are not found to be equivalent to others in the set are included as novel Ensembl-based GeneCards gene entries. These primary sources provide annotations for aliases, descriptions, previous symbols, gene category, location, summaries, paralogs and ncRNA details. Once the gene list is in place with these significant annotations, over 80 data sources, including those noted above and others (12,18,22,36,46,47) are mined for thousands of additional descriptors.
The data collection and integration process, which runs periodically (typically every 3–5 months) to ensure ongoing access to recent updates, culminates in producing an integrated database, which is available in plain text and XML files, as well as MySQL dumps.
The version 3 database
The GeneCards data model is complex. In legacy GeneCards Versions 2.xx, information is stored in flat files, one file per gene. Version 3 uses a persistent object/relational approach, attempting to model all of the data entities and relationships in an efficient manner. This allows diverse functions of displaying single genes, extracting attribute slices and performing complex queries for sets of genes, and performing well on both full text and field-specific searches. Since the information is collected by interrogating dozens of sources, it is initially organized on a source-by-source basis. However, the relational database structure also makes it possible to present the data to the users organized by topics of interest, e.g. with all diseases grouped together, irrespective of the mined source. The V3 database (Figure 10) consists of 84 entities and 28 relationships. These database objects are implemented as 112 tables and two views (data and system), interlinked by 87 foreign keys. The central entity is the ‘genes’ entity, with attributes that include symbol, GeneCards identifier and origin (HGNC, Entrez Gene or ENSEMBL). The data model parallels that of the web-card, with some of the intricate sections (e.g. gene function) represented by several tables. An object-oriented interface to the data is facilitated by Propel (48), an open-source Object-Relational Mapping (ORM) framework for the PHP programming language. Our V2 to V3 migration path uses the project’s XML files, already organized in both data source and functional presentations (2) as the source for populating the relational database. The complete schema is available upon request.
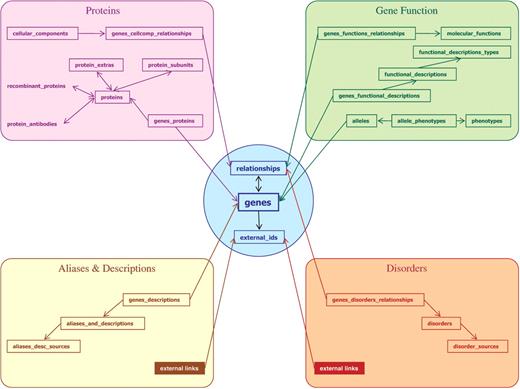
Sample of GeneCards Version 3 (revision 3.02) gene-centric relational database entities and their relationships, with associated web-card sections.
The new search engine
As described earlier, the new search engine is very fast. Much of the speed stems from the selection of the Lucene indexing technology (49) used also by sites like Wikipedia (50) and Microsoft’s Bing (51). We have chosen the Solr (52) server, which combines the Lucene library with XML, HTTP, hit highlighting and faceted navigation [a mechanism that enables a user to browse information along multiple paths (53)], enabling support for both field specific and full text searches, and having maturity, robustness and open-source availability. One shortcoming is that Lucene does not store information hierarchically, whereas GeneCards gene-specific data is by nature hierarchical. For example, the ‘Proteins’ section of the card contains information from UniProtKB/Swiss-Prot which in turn contains accession numbers, links to PDB structures, subunit information and so on. To produce optimal search results, both accurate and context-specific, the engine must identify the specific subsections of matched information in the relevant genes. However, all current, mature, search engines work at a higher level when identifying positive results; the only information that is returned by the search engine is the identity of the ‘document’ (e.g. specific gene in our case) that was found. Solr does offer the ability to return more detailed data, including a portion of words surrounding the hit, but in a flat form and without the hierarchical structure needed to enable identification of the specific subsection of the card that contains the query string. Often the same text phrase is found in dozens of places, making it impossible, in most cases, to identify which specific card sections or subsections (e.g. disorders and/or literature) define the complete hierarchical context of the hit. Our solution is to provide two-phased searches, enabled by two indexes, the primary index and the secondary index. The primary index is populated by an automatically generated flattened version of complete GeneCards textual information; the secondary index is populated with each individual sub-element as its own ‘document’, annotated with its associated genes. A typical work flow, say for a keyword search for cancer, is as follows: (i) Query the primary index to look for all gene records that contain ‘cancer’—to which the system quickly returns a list of ∼8000 genes, including MSH2. (ii) When the user requests to open the minicard for that gene, query the secondary index for a list of detailed database records, mapped to the relevant subsections of the card, which are associated with the gene MSH2 and contain the keyword cancer, and highlight the hits coherently and within context, in the minicard. Since this step is done upon demand, for a limited subset of the genes, valuable time is saved during the initial quest for matched genes. (iii) When the user requests to view the complete GeneCard for MSH2 by clicking on that symbol from within the search results, highlight the search term cancer in all places that it occurs in the card.
From an implementation standpoint, the GeneCards database relationships sometimes involve five or more joins, and there are several thousand relationship variations among the approximately 100 different tables. Consequently, preprocessing the data by maintaining optimized sets of typical queries is not feasible. The efficient indexes described earlier are built by a recursive crawler, which iterates over the relational table data structures associated with each gene, and discovers associated annotations. That data is categorized for faceted searching, and then transformed into valid virtual documents for the relevant index(es). When the database schema changes, say to accommodate new GeneCards data sources, the table-driven crawler code does not need to be modified. A challenge that was overcome was to ensure that the crawler would not enter infinite loops; this was achieved by carefully defining the terminal nodes in the network-like database schema. A major advantage of the crawler is its ability to create custom ‘perspectives’ automatically. GeneCards has traditionally been gene-centric in its organization of information. In V2, this was reflected in the underlying one gene per file technology. For the relational V3, other views of the data are naturally available. Without major changes to its architecture, the crawler is capable of re-rendering the nature of the GeneCards search to return hits that are not genes. One could then, for example, search for keywords (e.g. muscle) and receive hits presenting lists of associated disorders instead of (or in addition to) lists of associated genes. This will, one day, allow GeneCards to create new ways of presenting its rich data, without the need for major search-engine rewrites.
Version 2 infrastructure versus Version 3 infrastructure
V2 used the text cards as input for the web cards, for the GeneALaCart batch query system and for the Glimpse index (54) that served its searches. Version 3 uses its MySQL (55) database as input to its novel searchable word-set collection process, which provides input to a Lucene (49)-based search engine, and to drive the V3 GeneDecks partner hunting and set distillation tools. (56). V3 web-cards are currently implemented as a hybrid system, with the contents and user interface based on V2, but with the addition of a search bar that enables powerful V3 searches from each card.
Quality assurance and statistics via GeneQArds
GeneQArds is a resident quality assurance (QA) tool, enhanced in V3 to: (i) assess the integrity of the migration from the V2 text file system into the V3 MySQL database, and (ii) validate and quantify the results of the new V3 search engine. The GeneCards data transformation between versions is a multi-step pipeline which includes creating intermediate XML files as well as populating the large set of tables (Figure 11). To ensure exactitude, we have developed a mechanism, based on SQL queries and PHP modules, which builds a binary matrix indicating the presence or absence of source data for all gene entries. This matrix also serves as the foundation of the GeneCards site’s statistical graphs and its GIFtS annotation scores. This binary matrix is compared to its counterpart for the V2 text files, built with a set of V2 Perl quality assurance programs. The produced report provides a good initial assessment of the integrity of the database, and also points to the possible sources of errors. For example, paucity of V3 genes with annotation derived from a given source indicates a data mining setback, and provides a clue regarding the cause of error. This first-tier comparison alone is not sufficient, since the binary matrices do not contain details about each of the source-specific annotation fields (e.g. protein name, subunit and PDB identifiers within the Swiss-Prot annotation source). Therefore, a set of very detailed SQL queries are also run against the V3 database, with results compared against similar checks of the text cards, as well as previous version loads of the database. The search engine comparison tool enables version comparisons both by single query (via a web interface) and batch query (via command line) for all types of searches (keywords, symbols only, symbol/alias and mixed external identifiers). The results are summarized in a report, which includes the amount of time each search took, lists of distinct genes found by one of the search engines but not the other, and a list of genes found in both versions. To enable tracking the discrepancies, the report also notes the context for each of the hits (e.g. the keyword ‘cancer’ was found in gene TP53 in the proteins, disorders and summary sections) and provides a deep link for further scrutiny. The search engine comparison tool uses internal persistent MySQL tables which contain: (i) all single queries invoked by testers in GeneQArds, (ii) the 500 most frequent queries against the live GeneCards site, (iii) all queries that previously involved errors and (iv) the results of all comparisons. These tables help extend the power of GeneQArds, affording accurate multifaceted QA performance. One GeneQards output is a distribution of gene hits differences between two versions (Figure 12), allowing the tester to assess improvements or degradations. We have analyzed the trends of the results, followed by detailed inspection of ∼10% of the isolated anomalies. The fact that GeneQArds combines both white box (by taking advantage of knowing the internals of the system, e.g. by interrogating specific database tables) as well as black box (non-biased/external, e.g. by measuring hit counts) testing, will enable us to zero in on the remaining deficiencies. Many of the GeneQArds tools are available upon request.
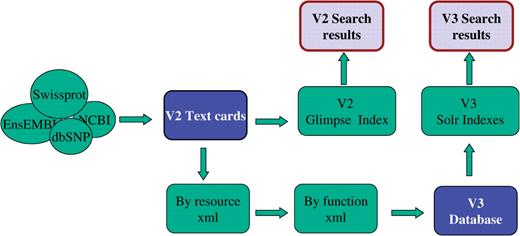
The V2 and V3 database collection/integration pipeline and search application flow.
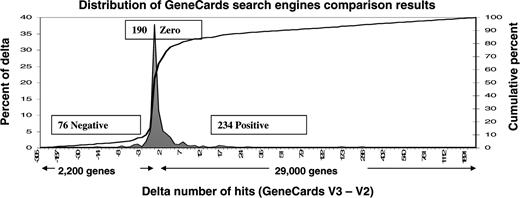
GeneQArds statistical comparison of GeneCards V2 and V3 search results, for each of the 500 most frequently searched terms in 2008, showing vast improvements for V3. The cases where V2 finds more hits reflect many V2 false positives, some V2 fields that haven’t as yet been incorporated into the V3 database, and some isolated anomalies that are still under investigation.
Supporting Software
GeneCards Version 2.xx is implemented in Perl, with indexing provided by the University of Arizona’s Glimpse software (54). GeneCards Version 3 uses XML, MySQL (with default fast MyISAM tables), and PHP, together with the Propel (48) Object-Relational Mapping (ORM) framework for PHP. The latter provides foreign key metadata in its configuration information, to compensate for MyISAM’s lack of support for foreign key constraints. V3 uses Smarty templates, and the Lucene (49) search engine powered by Solr (52). GeneDecks’s Partner Hunter and Set Distiller server is written in Java. GeneQArds for V3 is implemented in PHP and is fully integrated with the V3 MySQL database. The other components of the GeneCards suite are written in Perl and MySQL.
Applications, advantages and future directions
GeneCards and its suite of tools have been instrumental in several recent collaborative projects with a biomedical end-point. GeneCards’ contribution is detailed herein:
SYNLET—Regulatory control networks of synthetic lethality.
This EU-funded project (http://synlet.izbi.uni-leipzig.de/) addresses robustness of phenotypic function on the basis of Synthetic Lethality—as proposed for novel cancer treatment regimes. It derives novel concepts, methodologies and algorithms for annotation and analysis of regulatory networks, with focus on tumorigenesis and drug resistance. It aims at identifying key proteins of cellular escape mechanisms that overcome lethality of drugs and find ways to block them, utilizing siRNA. GeneDecks was one of the main tools which served the consortium to select candidates for the SiRNA experiments. Specifically, in the Partner Hunter mode, Synlet searched for partners for genes which underwent significant expression diminution in microarray experiments done on resistant and non-resistant neuroblastoma cell lines, seeking inactivation targets among partners to which the tissue has been ‘addicted‘. The Set Distiller mode of GeneDecks enables the identification of annotation descriptors shared by experimentally-obtained gene sets, allowing the assessment of their belonging to specific functional classes, hence a judicious selection of inactivation targets.
SysKid—Systems biology towards novel chronic kidney disease diagnosis and treatment
This EU-funded project focuses on chronic kidney disease, with diabetes mellitus and hypertension being the most prevalent causative conditions. It assumes that despite it being diverse in etiology, the underlying molecular pathophysiology of different manifestations of this condition may be similar. The project aims at obtaining an integrated view on the disease in the realm of systems biology, based on integrated analyses of high-throughput OMICS data. The GeneCards V3 database and advanced search engine are instrumental for the integration process and in identifying the most promising disease markers. As an example, GeneCards will be used to identify relevant genes for metabolomics pointers. In this framework, a ‘Genecards 100k’ effort is currently under way, aiming at increasing the number of Gene entries towards 100 000. This will be accomplished mainly through a significant expansion of GeneCards’ scope in the realm of non-protein-coding RNA genes, and resolving a large number of genes currently entitled ‘uncategorized’. In parallel, a project-specific database (GeneKid) will be created, to house incoming OMICs data in GeneCards-compatible tables, thus facilitating the systems biology analyses.
Research facilitated by GeneALaCart
GeneALaCart has contributed to numerous collaborative efforts, and, based on user feedback, has been helpful to hundreds of research groups. A point of strength is its capacity to do cross-database identifier mappings of genes and proteins, using the mixed identifier feature. GeneALaCart provides systematic and detailed annotation of gene lists, obtained, for example, from differential expression, transcriptional regulation, siRNA screens or genome-wide genetic association studies. Some users seek specific information for their gene lists, such as the elucidation of their potential drug targets, their orthologs in other species, or their annotated SNP lists. Many of the specific uses have clinical implications, for example assistance in the choice of SNPs relevant to complex diseases studies, integration of phenotype and genotype information in clinical patient information systems, or deciphering genes implicated in clinical studies, including brain disorders or immunity.
Research facilitated by GeneAnnot
An example of where GeneCards gene expression and annotation has been applied (at the University of Modena, Italy) is the development of a novel set of custom Chip Definition Files (CDF) and corresponding Bioconductor libraries for Affymetrix human GeneChips, based on information supplied by GeneAnnot (57).
Advantages of GeneCards V3 search results
We sampled fifteen single word queries, most extracted from our list of popular GeneCards search terms, and compared the number of hits found by GeneCards to those found for human genes by similar systems [NCBI Entrez Gene(5), Ensembl(6) and Harvester(58)]. GeneCards and Entrez Gene offer users the option to easily download the complete result set (in addition to the default paged behavior that all share). The average response times in seconds (±SD) were: GeneCards 5.5 (±0.63), Entrez Gene 2.9 (±0.59), Ensembl 1.9 (±0.59), and Harvester 2.5 (±0.51), with the GeneCards results offering detailed ‘minicards‘. In a majority of the cases, GeneCards finds more hits, due to the unique richness of its contributing data sources. For example, the search for EGFR finds: (i) a unique hit for the gene AGK because its functional information from Swiss-Prot (7) has the phrase ‘Overexpression increases the formation and secretion of LPA, resulting in transactivation of ‘EGFR’ and activation of the downstream MAPK signaling pathway, leading to increased cell growth’ and (ii) a hit for the gene A2 M because STRING (31) identifies a protein–protein interaction between the two genes. Similarly, the search for retinoblastoma in GeneCards finds a unique hit for AANAT due to its association with this disorder via Novoseek (33). Table 1 summarizes the benchmark’s details, and Figure 13 presents qualitative differences for these two specific examples. While assessing the quality of the extra hits (see Quality assurance and statistics via GeneQArds, in ‘Methods’ section), in addition to the largely positive results as demonstrated by these examples (right tail of the distribution, Figure 12), we did find that the search engine’s stemming is at times over-zealous. For example, when searching for ‘batten disease’ (without quotes), a false hit is found for the gene IL6, since one of its publications authors is named Battenfeld. We will delve into Solr’s stemming rules, improve our engine to not apply stemming to author lists, and continue to probe the QA results in depth as we enhance GeneCards in future versions.
Search benchmark: comparison of the total number of hits found when searching for 15 popular search terms (selected from the list of top 100 terms queried in GeneCards during 2008 and 2009) in GeneCards, Entrez Gene, Ensembl and Harvester
| Search term | Total number of hits (genes) | |||
|---|---|---|---|---|
| GeneCards V3 | Entrez Genea | Ensembl | Harvester | |
| Asthma | 861 | 426 | 35 | 42 |
| Estrogen | 1912 | 572 | 152 | 383 |
| Hippocampusb | 726 | 153 | 30 | 1197 |
| Olfactoryb | 1449 | 2755 | 1339 | 828 |
| Retinoblastoma | 643 | 209 | 38 | 29 |
| Tongue | 348 | 75 | 12 | 80 |
| VIMENTIN | 243 | 74 | 4 | 48 |
| BRCA1 | 651 | 312 | 23 | 143 |
| CDKN1A | 79 | 104 | 11 | 75 |
| CFTR | 264 | 145 | 30 | 54 |
| EGFR | 870 | 495 | 7 | 43 |
| GAPDH | 78 | 164 | 55 | 34 |
| MTHFR | 116 | 56 | 2 | 11 |
| TP53 | 884 | 419 | 26 | 401 |
| VEGF | 1012 | 512 | 9 | 61 |
aIncludes entries discontinued by NCBI—see examples in Figure 13.
bSearch terms which are not from the list of top 100 searches.
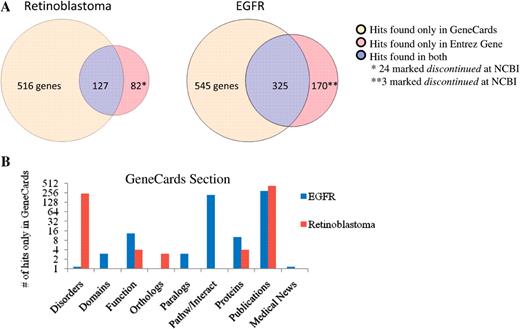
(A) Comparison of the number of hits (genes) found by GeneCards and Entrez Gene for two popular searches (EGFR and Retinoblastoma) and (B) the distribution of those hits within the various sections in GeneCards.
Future directions
In addition to specific enhancements and improvements directed by the above projects, we will continue to enhance GeneCards core features. An intriguing future challenge is to devise an algorithm for unifying disease names/descriptions and enable these tables to be merged. We’ would like to increase our pathway repertoire, and are considering adding public domain (e.g. Reactome) and/or additional commercial pathways. We hope to expand the ‘Function’ section to include animal models from species other than mouse.
For GeneLoc, we are considering incorporating UCSC and/or CCDS identifiers. We plan to migrate out of the currently implemented hybrid web-card system, with contents and user interface still based on V2, to web-cards that fully use the relational database and PHP/Propel infrastructure. In parallel, we will continue to expand and improve the GeneDecks algorithms.
Summary
GeneCards has evolved tremendously over the years, progressing from being an effective ‘one-stop shop‘ source of information for scientists’ particular human genes of interest, to becoming a facilitator for sophisticated systems-biology efforts. We envision that its updated functionality and new infrastructure will continue to provide an effective research and development platform for many years to come, and look forward to pursuing more adventures.
Funding
The Weizmann Institute of Science Crown Human Genome Center and the Phyllis and Joseph Gurwin Fund for Scientific Advancement; EU Specific Targeted Research Project consortium ‘Regulatory Control Networks Synthetic Lethality’ (SYNLET—EU FP6 project number 043312); EU Systems Biology towards Novel Chronic Kidney Disease Diagnosis and Treatment Project consortium (SysKid—EU FP7 project number 241544); Xennex, Inc., Cambridge MA. Funding for open access charge: The Weizmann Institute of Science Crown Human Genome Center.
Conflict of interest: None declared.
Acknowledgements
The authors thank the reviewers for crucial insights and suggestions that have helped improve this manuscript, Elena Matusevich and Yakov Perlman for their initial implementation of GeneCards Version 3, David Warshawsky for providing the model and data sources for highly-targeted reagents, Edna Ben-Asher and Orit Shmueli for defining the initial SNP filtering algorithms, Ido Zak for improving its implementation, Ohad Greenspan for implementing the alternative splicing diagram, and Liora Strichman-Almashanu for her mouse phenotype initiative, for pioneering GeneQArds, and for initial V3 data modeling work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments
We appreciate the positive review of GeneCards presented by Hisham M. Eldai!
In response to his question about the use of PHP and Propel as opposed to using Perl with Tangram, we'd like to emphasize that our first decision was to migrate from Perl to PHP. PHP was initially developed for Web applications, and is now widely used in that context, with a significant development community. We were at the point in the project where maintenance was becoming very cumbersome, and a major overhaul was in order. We wanted to make sure to avoid the temptation of adding new patches, and preferred to take the time to redesign based on a completely new and more suitable platform. We felt that since PHP provides several standardized platforms for object mapping, it would afford us a clean way of managing our data model and limiting code changes as the system evolved. So frankly, Tangram was never considered, since it's provided only for Perl and isn't lightweight, and we had already decided to migrate to using PHP. Once that decision was made, Propel was chosen because it offers a popular, well-maintained, standardized, strongly-typed ORM layer.
Marilyn Safran for the GeneCards team
Conflict of Interest:
None declared
Integration of different data sources allows for powerful mining outcomes and development of new technologies. I see that the GeneCards sets a true example of efficient databases and technology integrations. Preserving the atomic nature while consistently expanding to keep development going. Moreover the access to vendors links is just so precious both as a business model strategy and user-guidance.
I particularly admired the visualization solutions offered in terms of gene cards, expression graphs comparing normal and cancer tissue (especially the diamonds along the x-axis), splice variants offset coordinate recalculation and the color scheme to represent the entity type and the data-source.
However, I seek your response to me with regard to the choice of PHP Propel mapper since the earlier version of GeneCards used Perl and that language has a Tangram object relational mapper through its T2 module. So what drove the choice of Propel over Tangram?
Conflict of Interest:
None declared