





Abstract
The proliferation of biological databases and the easy access enabled by the Internet is having a beneficial impact on biological sciences and transforming the way research is conducted. There are ∼1100 molecular biology databases dispersed throughout the Internet. To assist in the functional, structural and evolutionary analysis of the abundant number of novel proteins continually identified from whole-genome sequencing, we introduce the PROFESS (PROtein Function, Evolution, Structure and Sequence) database. Our database is designed to be versatile and expandable and will not confine analysis to a pre-existing set of data relationships. A fundamental component of this approach is the development of an intuitive query system that incorporates a variety of similarity functions capable of generating data relationships not conceived during the creation of the database. The utility of PROFESS is demonstrated by the analysis of the structural drift of homologous proteins and the identification of potential pancreatic cancer therapeutic targets based on the observation of protein–protein interaction networks.
Database URL:http://cse.unl.edu/∼profess/
Introduction
There are ∼1100 molecular biology databases freely available to the public online (1,2). These databases constitute the extent of our knowledge related to genomics, proteomics, metabolomics, and structural genomics. Most serve as data warehouses with simple interfaces for data retrieval (3). To address more complex questions, biologists are routinely required to develop new databases by filtering information from existing databases (4). Even though this is extremely inefficient, there are a growing number of specialized databases designed around single topics. Unfortunately, this simply propagates the underlying problem: an inability to utilize the data outside the constraints imposed by the database designers (5). Capitalizing on the potential of biological information requires the development of a next-generation database that enables biologists to explore biological data in new ways. The key to solving this problem is to move the design focus from the database structure (predefined relationships between fields) to a fluid association that can be adapted to a biologist’s questions (6) without re-designing the underlying data structure. However, there are barriers to linking individual databases because of different data formats and structure (7, 8). Thus, it was essential to this effort to implement a new approach to integrate diverse biological databases (9).
Most of the work on database integration has focused on business and spatio-temporal data (10, 11). Satisfying, general and practical solutions have proven to be elusive for these complex data sources, which are actually simple compared to biological data. Nevertheless, the most versatile of the solutions is to use a separate adapter, or ‘wrapper’ (Figure 1), program around each source database (12). The ‘wrappers’ provide a simplified ‘view’ of the source database presented in a form that is easier-to-use than the original source database. In fact, some parts of the source data may be completely omitted in this repacked presentation, leaving only the parts of the data that are needed for the enterprise that wants to use it. The advantage of the ‘answering queries using views’ approach to the database integration problem is that it reduces the integration problem to two steps: (i) building wrappers of the source databases, thereby providing simple ‘views’, and (ii) applying standard database queries on the views. Thus, implementing wrappers enables a robust query system that incorporates a variety of similarity functions capable of generating data relationships not conceived during the creation of the database. This will allow the user to move beyond simple text-based queries. Therefore, the PROFESS (PROtein Function, Evolution, Structure and Sequence) database uses wrappers to assist in the structural, functional and evolutionary analysis of the abundant number of novel proteins continually identified from whole-genome sequencing.
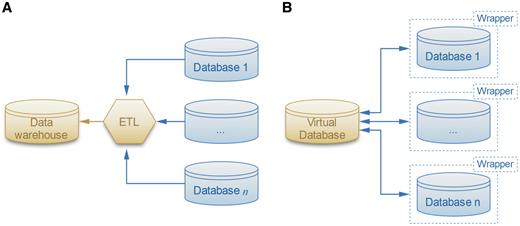
Two solutions for the data integration problem. (A) The ETL software extracts, transforms and loads the data sources into the warehouse. (B) The more flexible local-as-view method defines a virtual database that interacts with data sources through wrappers, which provide simplified views of the original databases.
Database content
Fourteen sources of data were integrated to create PROFESS (Table 1) using a local-as-view (LAV) modular approach (Figure 1B) (see the ‘Method for data integration’ section for details). The modular functionality of PROFESS coupled with user friendly searching capabilities makes PROFESS particularly useful for asking a range of questions about the sequence, structure, and functional relationship of evolutionary and functionally related proteins. A user interacts with PROFESS through a web interface using a functional-style query language that is translated to the structure query language (SQL) for mining PROFESS (Figure 2A). The core of PROFESS established a relationship between the Protein Data Bank (PDB) (13) and the eggNOG databases (14, 15) (Figure 2B). The link between eggNOG with the PDB was established using the proteins UniProt accession numbers and the UniProt Mapping service (16).
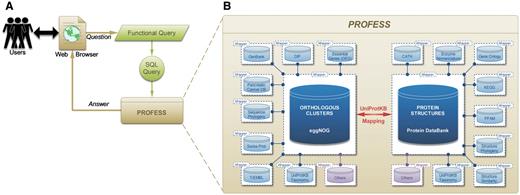
Outline of the PROFESS database. (A) The relationship of the user interface to the functional query system (green) to the PROFESS databases; and (B) the core databases integrated in PROFESS. The central eggNOG-PDB linkage is shown in red, double arrows indicate intensive interactions, blue boxes represent databases available on the internet, and purple boxes denote other databases to be integrated in the future. Each additional data set interacts with the PROFESS core through the use of wrapper programs to make query language uniform.
Core databases currently integrated in PROFESS
| Name | PROFESS level | Link | Reference |
|---|---|---|---|
| CATH database | Structure | http://www.cathdb.info/ | (27) |
| eggNOG database | Function | http://eggnog.embl.de/ | (15) |
| Enzyme classification | Function | http://www.chem.qmul.ac.uk/iubmb/enzyme/ | (19) |
| Database of essential genes (DEG) | Evolution | http://www.essentialgene.org/ | (26) |
| Database of interaction proteins (DIP) | Function | http://dip.doe-mbi.ucla.edu/ | (22) |
| Orthologous structure and sequence-based phylogenies | Evolution | This database | |
| Orthologous structure similarity comparisons | Structure | This database | |
| Pancreatic cancer related proteins | Disease | This database | |
| Gene ontology | Function | http://www.geneontology.org/ | (18) |
| GenBank | Sequence | http://www.ncbi.nlm.nih.gov/Genbank/ | (60) |
| KEGG ligands | Function | http://www.genome.jp/kegg/ligand.html | (20) |
| Protein data bank (PDB) | Structure | http://www.rcsb.org/ | (13) |
| Protein families (PFAM) database | Function | http://pfam.sanger.ac.uk/ | (17) |
| Protein/protein interactions in E. coli | Function | http://genome.cshlp.org/content/16/5/686.abstract | (21) |
| SCOP | Structure | http://www.bio.cam.ac.uk/scop/ | (28) |
| Swiss-Prot | Sequence | http://www.uniprot.org/ | (61) |
| TrEMBL | Sequence | http://www.uniprot.org/ | (61) |
| UniProtKB taxonomy | All | http://www.uniprot.org/taxonomy/ | (16) |
To simplify the interface, each orthologous protein family has four tabs containing information about: function, evolution, structure and sequence. An additional tab, diseases, shows linkages between human proteins and information culled from databases devoted to the functional genomics and proteomics of particular diseases. Each protein is annotated with its source organism using the UniProtKB taxonomy database (16). Each level of the PROFESS database mines pieces of information from all the integrated databases and provides the user with comprehensive tables highlighting annotations (Figure 3). The tables are defined as independent modules, each providing a unique representation of the integrated data. Each module can be activated or deactivated, depending on the specific needs of the user. PROFESS is not limited in the size or type of data that can be incorporated due to the LAV approach coupled with a modular interface. This allows the integration of biological data for rapid identification of biologically relevant similarities or differences between various protein functions.
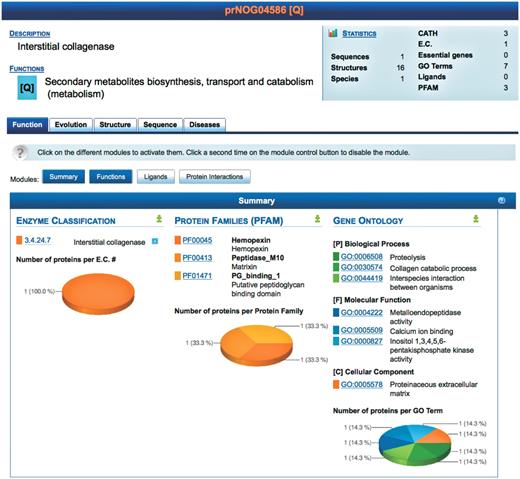
Screenshot of the result page for prNOG04586. A brief description of the cluster is displayed (top) along with statistics. Detailed data is shown for each level (function, evolution, structure, sequence and disease). At each level, data is further clustered into different modules, each module providing a unique view of the data. Each module may be activated or deactivated depending on the needs of the user. The screenshot shows the module summarizing functional annotations of proteins in prNOG04586. Data is mined from the enzyme classification, the protein families database and the gene ontology. For each database, PROFESS shows entries related to proteins within cluster prNOG04586. The pie charts represent the relative frequency of each database entry within the orthologous cluster.
Function
The Function tab of PROFESS summarizes the biological function of an orthologous cluster. For three primary descriptions of protein function, the numbers of proteins within each class (within the current orthologous cluster) are computed and the distributions are represented as pie charts. This allows the user to quickly differentiate relevant classes from outliers. Classes are sorted by decreasing number of proteins. The darker the color in the pie chart, the higher the number of proteins. As an example a search of ‘collagenase’ retrieved 34 different orthologous groups, one group (prNOG04586) is shown in Figure 3.
The Function tab also contains three unique sub-modules that describe the primary biological function of a cluster of orthologs. The first module, Functions, is a table of the functional annotations for a protein structure taken from the PDB, including the protein families (PFAM) (17), gene ontology (GO) (18) and enzyme commission (EC) number (19). It is left to the user to examine the combination of annotations to assess its overall consistency and to identify possible mis-annotations. Protein function can also be described by protein interaction partners, therefore two additional modules (ligands and protein interactions) list the ligands and proteins experimentally shown to interact with members of the eggNOG family. The Ligands module displays details about ligands known to bind a protein based on ligand bound structures in the PDB as well as cross-references to the Kyoto Encyclopedia of genes and genomes (KEGG) (20). Common buffers, detergents, ions and solvents are listed separately to provide rapid access to biologically relevant data. The protein interactions module lists protein interactions found in Escherichia coli (21). The interactions were correlated to the corresponding PDB ID by matching bait and prey genes to their representative eggNOG cluster. The protein interactions module also integrates the 69 171 manually curated protein/protein interactions (as of April 2010) in 274 organisms from the database of interacting proteins (22).
Evolution
The Evolution tab of PROFESS displays a table of essential genes, along with sequence- and structure-based phylogenetic trees. The sequence tree shows the unrooted phylogenetic tree created from the tree files downloaded from the eggNOG database (14, 15). The final image was generated using DrawTree from the Phylip package (23, 24). The sequence trees contain many branches and nodes and provide an overview of the overall bushy nature of the cluster, a more detailed tree can be found by searching a particular cluster using the eggNOG database (14, 15).
The structure tree shows the unrooted phylogenetic tree generated using PDB protein structures. The structures were aligned using MAMMOTH-mult (25) and the structure based sequence alignment was used to compute the trees and image. Branch lengths for each structure alignment from MAMMOTH-mult (25) were measured by our in house software and minimized using the neighbor joining program implemented in Phylip (24). The final image was generated in the same manner as the sequence tree.
The essential genes module of the evolution level shows whether the protein in the orthologous cluster is essential and was obtained from the database of essential genes (DEG) (26). As of version 5.4, DEG includes 5260 essential prokaryotic genes and 5040 eukaryotic genes extracted from the literature. Genes are displayed with corresponding protein structures from the PDB (see module Sequence similarities for more details about the association gene/structure). As with all databases, DEG should not be viewed as an exhaustive or complete list of all essential genes, but only as a work in progress. For instance, well-established and obviously essential genes may not be included in DEG, because its focus is on the current literature. Since PROFESS is continually updated and expanded, the list of classified essential genes will continue to expand as new studies are carried out and as DEG reaches deeper into the older literature.
Structure
The structure tab of PROFESS contains all structures associated with an eggNOG cluster and is linked together by their Uniprot accession numbers. Therefore, the availability of a structure in PROFESS is limited to a preexisting Uniprot-eggNOG linkage. If a Uniprot-eggNOG linkage does not exist for a queried structure, then the structure is not present in PROFESS and will not be displayed in the results summary. The structure tab also contains an aggregate table of data from the CATH (27) and SCOP (28) databases. Due to copyright restrictions, links are provided to retrieve data from the SCOP website rather than reproducing SCOP data on our pages.
The structure tab is designed to ease searching for all orthologous clusters with a particular fold. This is accomplished by either direct or iterative searching for a particular CATH ID number. The direct searching method would be to enter a known CATH ID into the PROFESSor to find the correlated orthologous clusters. In iterative searching, a user first searches for a protein structure with the PROFESSor to identify the orthologous group, finds the CATH ID in the structure tab, and then searches the selected CATH ID with the PROFESSor. Both searching methods will generate a list of orthologous clusters that contain the protein fold of interest.
The structure level also contains all pairwise structure alignments of an orthologous cluster. The pairwise structure comparison tool DaliLite (29, 30) was used to measure the backbone structure similarity of proteins within each orthologous cluster defined by the eggNOG database. All-against-all pairwise structural comparisons were carried out for all 224 847 NOGs with 401 967 total structure comparisons. Structure calculations were completed with help from the Holland Computing Center of the University of Nebraska-Lincoln.
The Dali Z-scores were normalized to calculate a fractional structure similarity (FSS) score: FSS = ZAB/ZAA, where ZAB is the Dali Z-score when protein B is compared to protein A and ZAA is the Z-score when protein A is compared to itself. Thus, ZAA represents the maximum Z-score that can be achieved for perfect similarity. FSS provides a simple normalized and quantitative measure of the distance the two proteins have diverged in their structures.
Sequence
The sequence tab of PROFESS lists all protein sequences within the orthologous cluster. The sequence tab also provides the Uniprot accession numbers, molecular weight, length of sequence and when available the structure. A list of all sequences from each orthologous group is downloadable into FASTA format and each sequence can be individually copied and pasted into a text document in FASTA format.
Diseases
The diseases tab of PROFESS is reserved for gene and protein information identified throughout the literature as being involved in various human diseases. Currently, PROFESS includes information about genes and proteins involved in pancreatic cancer but this level of PROFESS will grow rapidly as new data is incorporated.
Query system for data mining
The PROFESSor
The primary search function of PROFESS is the PROFESSor (Figure 4), a unified text field that will assist the user to easily refine complex queries by dynamically suggesting entries from any integrated database. The PROFESSor assists the user by correcting for spelling errors using Levenshtein metrics, as well as providing a user defined focused browsing feature. For instance, upon typing in the query ‘collagenase’, the PROFESSor returns a drop down list of protein folds and functions that have known relation with collagenase (Figure 4). If a user selects the fold (CATH) suggestion, PROFESS will return all functional clusters known to contain that fold. The PROFESSor searches all other data sources within PROFESS in the same manner. In a single search, for example, the user can identify other protein functions with the same fold, similar ligands, or cellular localizations.
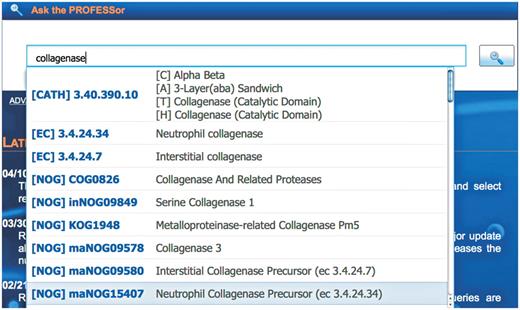
The PROFESSor query system. The PROFESSor is a dynamic search tool generated from the core databases to help the user to refine complex queries. Using the PROFESSor users are given suggestions for extending their search words/phrases that helps them rapidly and accurately find all functional, structure and sequence information about a particular protein and its relation to other protein functions, folds or ligands.
Advanced query system
Although the default views aims to provide a broad overview of protein functions, evolution, structures and sequences, users may need to create their own module—or view—to mine only those pieces of data required to answer a specific query. New views can be easily implemented using SQL queries, which give users full access to any data integrated within PROFESS. An example of an SQL query is shown in Figure 5A and is discussed below in the Applications section. The entity-relationship diagram describing the structure of PROFESS is provided in the online documentation and will help users to design the SQL queries. Like other modules, the data displayed in the custom view can be sorted and clustered as needed. The data can also be downloaded in CSV format for further analysis.
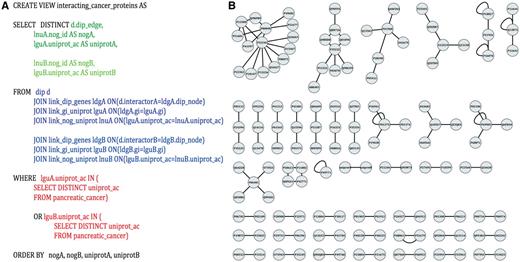
Identification of potential pancreatic cancer drug targets. (A) An example SQL query used to parse PROFESS to generate protein–protein interaction networks between pancreatic cancer-related proteins. Select (green) only the information relevant to solve the stipulated question from the dynamic join of relevant views from PROFESS (blue). The results are then filtered to mine only interactions involving proteins of interest (red). Parts of the query related to the first interactor are shown in darker colors, whereas sections of the query related to the second interactor are shown in lighter colors. (B) The SQL query on PROFESS resulted in a list of protein-protein interactions among the set of pancreatic cancer-related proteins. The interaction networks were displayed using Cytoscape (55). Identifying proteins that are part of a larger network provides one method to prioritize potential therapeutic targets among the set of pancreatic cancer-related proteins.
Functional-style query system
A fundamental component of PROFESS queries is to enable the users to incorporate a variety of new functions, which take as input a set of parameters and give as output a well-defined value or set of values. Such user-defined functions arise naturally in many applications. For example, we defined the CPASS similarity function that is capable of generating novel data relationships between proteins based on a sequence and structure similarity in ligand-binding sites (31). As another example, one may query for a relationship between the PFAM and the eggNOG databases, even though this relation is not explicitly defined in the PROFESS database. The first atomic function to be integrated is BLAST, which will be added shortly. It will enable users to retrieve orthologous clusters of proteins related to a protein sequence of interest. Input sequences will be aligned against all sequences from the eggNOG database. NOG clusters corresponding to significant hits will then be returned to the user. By providing a library of standard atomic functions, such as BLAST, the users will be able to compose the atomic functions in complex functional-style queries. A functional-style query is defined as a pipeline of any of the atomic functions, where the output of a function serves as input of the next function in the pipeline. The full description of the current set of functions in PROFESS will be available in the online documentation.
Method for data integration
Traditional data integration methods involve data warehousing, where the database extracts, transforms and loads (ETL) data from various sources into a single schema that is easy to query (Figure 1A). However ETL methods lack flexibility because they require the warehouse schema to be tightly coupled with the data sources. As a result, integrating new data sources requires considerable effort as the entire warehouse and subsequent queries need to be redefined. The warehouse schema may also have to be redesigned if one of the data sources schema changes after an update.
LAV method
To address the flexibility issues of widely-used ETL methods, the PROFESS database was designed using a flexible LAV method (12, 32) as shown in Figure 1B. LAV methods involve wrappers that provide an abstraction layer for each data set. Wrappers are software that translate the data sources and provide an abstract, simplified view of the integrated data sources. Although there have been prior integration efforts of structural data and functional data sources, the PROFESS system has a unique approach. It creates two internal wrappers, one for the integrated functional data and another for the integrated structural data. Then, it applies novel functions for the association between these two wrappers. This multi-step integration approach first merges the easier-to-integrate data sources, and then merges the harder-to-integrate data sources. Incomplete and incorrect information in the data source is one of the major difficulties with data integration. By first merging together closely related data sources, our method increases the likelihood that data from different sources will complete and correct each other. All of the annotations are reported to the user who can then use them to assess possible mis-annotations. In this way, PROFESS will help users overcome such problems as incomplete and misleading data annotations. Structural and functional data are often difficult to integrate because of different identification numbers, different functional definitions, and the absence of a direct link between the two data sources. Our multi-level integration approach first links all intermediate information to either the central functional wrapper (as defined by the eggNOG database) or to the central structural wrapper (as defined by the PDB database). The PDB-eggNOG bridge then serves as the intermediary for linking the functional and the structural wrappers. If this linkage does not exist, then the protein is not included in PROFESS.
The final step to achieve our flexibility and extendibility goals was to normalize our database structure. Database normalization was introduced by Codd in 1970 (33). It is a systematic process to ensure that a database structure will not be subject to anomalies after insertion, update, and deletion, that could lead to a loss of data integrity (34). Data normalization is also useful to reduce the need for restructuring the collection of relations as new types of data are introduced. There are currently five normal forms. The higher the normal form, the more robust the database structure is against inconsistencies. PROFESS was designed using the fifth normal form proposed by Fagin (35). The resulting entity-relationship diagram is shown in Figure 1.
However, selective denormalization was subsequently performed for performance reasons (36). In particular, the PROFESSor queries data from the table precalc_professor includes pre-computed joins between relations instead of using a dynamic view. To maintain data consistency, routines were implemented along with the wrappers to regenerate this table whenever new data is inserted into PROFESS.
Applications
Homologous protein structure comparison
PROFESS was initially created to test the hypothesis that proteins experience uniform structural drift following the divergence from a common ancestor. The goal of this effort was to address an apparent paradox in structural biology. Protein structures are generally considered invariant to maintain function (37), but sequence determines structure and sequence changes are the major determinant of evolution (38, 39). Therefore, what is the impact to a structure as a protein’s sequence undergoes genetic drift? Answering this question is conceptually straight-forward and simply required the structural comparison of functionally identical proteins from different phyla. Since the PDB is richest in bacterial proteins, functionally and evolutionarily similar protein structures from the two most populated bacterial phyla, Proteobacteria and Firmicutes, were the obvious choice. Thus, a key component of this analysis was the identification and extraction of Proteobacteria and Firmicutes protein structures from the PDB with an identical functional classification. Since the PDB is a classic example of a warehouse database with limited query capabilities, it was not possible to obtain this information directly from the PDB, and was our impetus to develop PROFESS. PROFESS was then used to associate PDB structures with both the eggNOG (evolutionary genealogy of genes: non-supervised orthologous groups) and phyla classifications. From this dataset, we identified 281 unique NOGs that contained a minimum of two Firmicutes organisms and two Proteobacteria organisms with a total of 3047 bacterial proteins (1066 Firmicutes and 1981 Proteobacteria). This set was subjected to a pairwise structural comparison between Proteobacteria–Proteobacteria structures, Firmicutes–Firmicutes structures and Proteobacteria–Firmicutes structures. The result was a greater difference between the Proteobacteria–Firmicutes structures, consistent with the ancient split between the two phyla. The results were incorporated into the PROFESS database.
Identification of potential pancreatic cancer therapeutic targets
Pancreatic cancer has the lowest five-year survival rate (5.5%) among cancers and is the fourth leading cause of cancer death in the USA (40, 41). Only three drugs have been approved by the FDA to treat pancreatic cancer, 5-fluorouracil (42), gemcitabine (43) and erlotinib (44), where these drugs are generally minimally effective and do not significantly prolong life (45). Thus, real progress in treating pancreatic cancer requires the identification of truly novel, yet druggable protein targets (46). One approach is to advance existing genomics and proteomics studies that populate the literature. Capitalizing on these existing data sets may provide a mechanism to identify potential drug discovery targets. Five separate proteomic studies have classified a total of 802 unique proteins that were differentially expressed in various pancreatic cancer cell lines (47–51). Similarly, a recent genomics analysis of mutation frequency rates in 24 pancreatic cancer cell lines identified 1331 genes with at least one genetic alteration (52).
To demonstrate the ease with which new data can be integrated into PROFESS and the flexibility of PROFESS to identify previously unknown relationships, PROFESS was used to test the hypothesis that the proteomic and functional genomics analysis of pancreatic cancer cells can be used to identify potential drug discovery targets. Even though changes in the expression profiles or a high mutation rates are not sufficient to verify that the protein is disease-related or therapeutically important (53, 54), it is possible that the discovery of protein–protein interactions networks could very well lead to possible drug targets among the dataset of pancreatic cancer-related proteins.
The manually curated pancreatic cells ‘omics’ data (PCOD) was integrated into PROFESS by implementing a wrapper and creating a new relationship in the database. The first issue addressed was that PCOD entries were identified by UniProt IDs, but the genes from the database of interacting protein (DIP) are identified using GIs. Using a standard ETL method would have required a program to create a new table that contains data from PCOD, DIP and the mapping between the UniProt IDs and GIs. Similar tables would have to be created for any additional relationship of interest to PCOD, which would led to an exponentially growth in the number of tables. Instead, our PROFESS database can take advantage of any data that has already been integrated into the database. Specifically, the UniProtKB mapping between UniProt IDs and GIs can be used in SQL queries to create new dynamic views. In this manner, PROFESS was mined to generate the view kog_interacting_cancer_protein, functional clusters of interacting pancreatic cancer-related proteins using the SQL statement shown in Figure 5A. The protein interaction network was quickly visualized (Figure 5B) by importing the output of the PROFESS SQL query into Cytoscape (55). Once a view has been created by a user, it will be automatically updated whenever relevant tables storing data from DIP and PCOD are updated. The resulting protein interaction networks illustrate the rapid data analysis that can be achieved using a fully integrated and flexible database based on protein function and structure. Using our LAV-based approach, the view for functional clusters of interacting pancreatic cancer-related proteins was obtained in less than four hours. Obtaining an equivalent table using the ETL method would have required a significant amount of additional effort.
Data access
PROFESS is freely accessible through the URL http://cse.unl.edu/∼profess and through our web-site http://bionmr-c1.unl.edu/. Data can be downloaded as parseable files in comma separated values (CSV) format from the web-interface or using RESTful HTTP requests that may be batched in scripts. Sequences and phylogenetic trees can be downloaded in FASTA and PHYLIP formats, respectively.
Implementation
The PROFESS database relies on the MySQL database management system. Wrappers are implemented in Java 1.6 and are platform independent. The web-user interface is implemented in PHP, Dynamic HTML, and the general Asynchronous Javascript and XML (AJAX) frameworks developed by Yahoo! (http://developer.yahoo.com/yui/) and ExtJS (http://extjs.com). PROFESS is running under Open SuSE Linux 11.0 on our new SunFire x4600 server, which features 8 AMD quad-core processors (32 cores) and 64 GB of memory.
Future directions
The initial implementation of PROFESS has focused on data integration and the development of basic searching capabilities. The future development of PROFESS will focus on the implementation of more robust user-friendly searching capabilities to augment the PROFESSor and SQL queries. Also, we will continue to expand PROFESS by the addition of other databases that contain information relevant to the structure, function and evolution of proteins and their association to human diseases. The identification of functional relationships depends on this essential information, where our new similarity and searching capabilities are expected to make associations not readily apparent within the original datasets. Additionally, to create a robust tool for functional annotation, the CPASS database (56) and results from functional screens of novel proteins by the Functional Annotation Screening Technology by NMR (FAST-NMR) (57, 58) will be integrated into PROFESS. Finally, PROFESS provides a great opportunity as the source data for many recent novel data mining and data classification algorithms that are especially designed for large-scale biological data (59).
Funding
This work was supported in part from the National Institute of Allergy and Infectious Diseases (grant number R21AI081154) to R.P. as well as by grants from the Nebraska Tobacco Settlement Biomedical Research Development Funds to R.P.; a Nebraska Research Council Interdisciplinary Research Grant to R.P.; a Milton E. Mohr Fellowship to T.T.; and a Fulbright Scholarship to P.R. The research was performed in facilities renovated with support from the National Institutes of Health (grant number RR015468-01). Funding for open access charges: National Institute of Allergy and Infectious Diseases (grant number R21AI081154) to R.P.
Conflict of interest. None declared.
The structure comparison work was completed utilizing the Holland Computing Center of the University of Nebraska-Lincoln. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases.
References
Author notes
†Present address: Thomas Triplet, Department of Computer Science, Concordia University, Montreal, Qc H3G-1M8, Canada.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}