





Abstract
Although most web-based biological databases (DBs) offer some type of web-based form to allow users to author DB queries, these query forms are quite restricted in the complexity of DB queries that they can formulate. They can typically query only one DB, and can query only a single type of object at a time (e.g. genes) with no possible interaction between the objects—that is, in SQL parlance, no joins are allowed between DB objects. Writing precise queries against biological DBs is usually left to a programmer skillful enough in complex DB query languages like SQL. We present a web interface for building precise queries for biological DBs that can construct much more precise queries than most web-based query forms, yet that is user friendly enough to be used by biologists. It supports queries containing multiple conditions, and connecting multiple object types without using the join concept, which is unintuitive to biologists. This interactive web interface is called the Structured Advanced Query Page (SAQP). Users interactively build up a wide range of query constructs. Interactive documentation within the SAQP describes the schema of the queried DBs. The SAQP is based on BioVelo, a query language based on list comprehension. The SAQP is part of the Pathway Tools software and is available as part of several bioinformatics web sites powered by Pathway Tools, including the BioCyc.org site that contains more than 500 Pathway/Genome DBs.
Introduction
Biological databases (DBs) now number in the hundreds, and are widely viewed as an essential part of post-genomic molecular biology. However, significant barriers limit biologists' access to biological DBs. Existing easy-to-use web-based query interfaces to biological DBs severely limit the complexity of queries that the user can formulate. Users who want to formulate complicated queries must learn both a DB query language such as SQL, and a computer programming language such as C or Java in which to embed those queries and process the results. Learning such languages is time consuming at best, and often presents an insurmountable hurdle for the biologist. One reason is that the semantics of SQL is based on concepts not commonly taught to scientists in the course of their University education (e.g. join).
We present a flexible web interface through which biologists and bioinformaticists can author precise queries to biological DBs. The queries that can be written with this interface, called the ‘Structured Advanced Query Page’ (SAQP), can be as precise as what can be expected from a computer programmer using expressive DB query languages like SQL. But the web interface can be used without programming expertise, and use of the SAQP avoids the types of errors that typically occur when writing computer programs—greatly reducing the barriers to writing precise queries.
A ‘precise query’ is formulated in such a way that it returns what the user wants without superfluous results. To enable precision in a query, an appropriate set of relational operators, as well as direct access to the underlying data, must be provided. A precise query is unlike an imprecise query that returns many results that must be disregarded by the user. A precise query can be simple or complex since if a user expects all the proteins of a DB, this can be done using a simple query whereas all proteins that are products of genes located in specific parts of the genome is a complex query requiring at least two classes of objects and several constraints.
A ‘complex query’ might involve more than one DB class, might include several DBs and specify many constraints, for example, ‘find all metabolic pathways containing more than four reactions for which all enzymes in the pathway are monomeric’ and ‘find all biochemical reactions that convert a carbohydrate to a phosphorylated carbohydrate, and where the molecular weight of the carbohydrate is less than 100’.
A ‘simple query’ typically involve searching one DB and one class, using one or two constraints. Examples include a query that searches for genes by name, or that searches for biochemical reactions by EC number, or for chemical compounds by molecular weight.
The flexibility, ease of use and precision of the SAQP are due to (i) ‘readable’, interactive, expandable form constructs for specifying search constraints and object attributes; (ii) the inclusion of variables in searches, which allow search components to refer to one another; and (iii) the inclusion of multiple search components within one query. The notion of readability of precise database queries is also presented in the doctoral dissertation of M. Bada (1).
The SAQP web interface is based on a DB query language newly developed as part of this project called BioVelo, that is more expressive than SQL, but that has a succinct syntax and a simpler semantics. In fact, the layout of the graphical interface of the SAQP is based on the syntax of BioVelo. We consider BioVelo's syntax terse enough to provide a user interface for direct entry of BioVelo queries. This interface, called the Free Form Advanced Query Page (FFAQP), is accessible from the SAQP in one click.
Figure 1 illustrates the general architecture of our DB query system. Two Web page interfaces are provided: the FFAQP and the SAQP. In this article, we focus on the SAQP.
The development of the SAQP was motivated by the need to provide biologists with the ability to query the large collection of Pathway/Genome DBs (PGDBs) being developed by users of the Pathway Tools software, including the more than 500 PGDBs within SRI's BioCyc collection (2,3) and the more than 200 PGDBs developed by groups outside SRI, such as YeastCyc and MouseCyc (4) (see BioCyc.org for a partial list). Pathway Tools PGDBs are managed using a Frame Knowledge Representation System called Ocelot (5). Ocelot is essentially a Common Lisp-based object-oriented database management system (DBMS) that uses a relational DBMS as a persistent back end. However, the relational aspect of Ocelot is invisible to the Ocelot user.
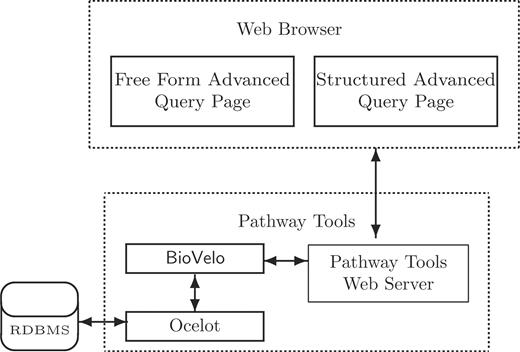
Two web page interfaces for constructing BioVelo queries interact with the Pathway Tools web server that communicates with a BioVelo query processor. Ocelot is an object-oriented DB system that can use a relational DB back end. The FFAQP and the SAQP are accessible at the web site BioCyc.org/query.shtml.
In summary, BioVelo serves as a database query language, currently built on Ocelot, and the SAQP is a user-friendly interface built on BioVelo. The BioVelo and SAQP implementations are applicable to any DB built using Ocelot, and thus generalize beyond Pathway Tools DBs such as the BioCyc DBs. Actually, the overall approach used for the SAQP is applicable to other relational and object-oriented DBs as BioVelo can be ported to other relational and object-oriented DBs.
This article is structured as follows. ‘Simple SAQP queries’ section presents the basic elements of the SAQP through a simple query with a few simple atomic conditions. ‘Pathway Tools schema’ section presents basic concepts of the Pathway Tools schema and how the SAQP interface can be used to explore and learn a schema to create precise queries. ‘Complex SAQP queries’ ‘Queries with several components’ and ‘Variables’ sections present more complex queries that illustrate the novel power of the SAQP. ‘Implementation’ section describes the implementation of the SAQP and the next section describes its limitations. The next following section describes related work. The final section presents the results of user evaluation of the SAQP. The Appendix gives a brief overview of BioVelo.
Simple SAQP queries
We introduce the SAQP by describing how to construct simple queries involving a single DB class, and how to specify the format of the query results. The BioCyc SAQP can be found at biocyc.org/query.shtml.
Step 1: select database and class
The first step in building a query is to specify at least one DB and the class of objects to search. Figure 2 shows an example of using the SAQP to query the class of protein monomers (Polypeptides) in the EcoCyc DB. Only one search component is used in this example. ‘Queries with several components’ section will show an example with multiple search components.
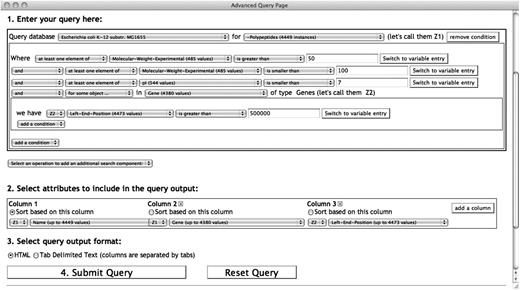
A SAQP query searching for EcoCyc (Escherichia coli) polypeptides constrained by experimentally determined molecular weight, isoelectric point and their genes. A three-column output table is specified containing the polypeptide name, gene name producing this polypeptide and the left-end-position of the gene on the genome. The atomic condition with a quantifier ‘for some object…’ retrieves only the polypeptides that are produced by genes located after the first 500 kb of the genome. Notice the use of two variables Z1 and Z2 in the query and in the output specification. The Z2 variable comes from the quantifier applied to the genes, whereas the Z1 variable comes from the search component main search objects, the polypeptides.
Step 2: specify conditions
Most queries include one or more conditions on the desired objects within the class. By clicking the button labeled add a condition in the initial blank SAQP, a ‘where’ clause is added—visually boxed—in the search component. This operation adds a selector for an ‘attribute’ (e.g. name) of the objects and a selector for a relational operator (e.g. contains the substring). It also adds a ‘free text box’ to enter a number or string. Several other relational operators are provided, such as is equal to, is not equal to and is a substring of. Regular expression matching is also available as an operator.
This new field forms an ‘atomic condition’. Additional atomic conditions can be added to the query by using the button labeled ‘add a condition’.
When clicking the drop-down selector for a relational operator, the list of relational operators provided is compatible with the type of the selected attribute. In the case of the attribute name, the selectable operators are for strings since the ‘type’ of the attribute name is string. This notion of type extends to all biological objects such as genes, proteins, metabolic pathways, reactions and compounds. Thus, the user cannot select an operator incompatible with the attribute of a class. The query in Figure 2 has added three atomic conditions to filter the selected polypeptides.
Step 3: define query results
The section titled Select attributes to include in the query output allows the user to describe the contents of the query results by selecting the attributes to display for each result object. The result of a query is always a table of at least one column. The tables have zero or more rows, one for each query result and each column is a selected attribute. A new column can be added by clicking the button add a column. A column can be removed by clicking its x icon. Each specified column will be generated in the resulting output table.
The selector provided in each column contains the list of accessible attributes for the object class selected for this query. When only one search component has been specified in the query and no subqueries with a quantifier are used, which is the case for Figure 2 (‘Queries with several components’ section describes search components in more detail), only one type of object is accessible. When more than one search component is specified or a subquery is used with a quantifier, a variable selector is provided to select the desired variable. The interface provides the number of possible objects having at least one value for each attribute.
The output table produced by the SAQP can be formatted in two possible styles: tabulated and HTML. For the tabulated format, no HTML is generated in the output result, and column entries are separated by a tab. It can be used as input to software such as Excel. For the HTML format, which is the preselected format, the generated output contains HTML tags for URL links and other visual formatting. It is the preferred format to navigate and analyze the results using a web browser.
The rows of the resulting table can be sorted based on any user-selected column. It can be resorted at will on any column in the output page as seen in Figure 3.
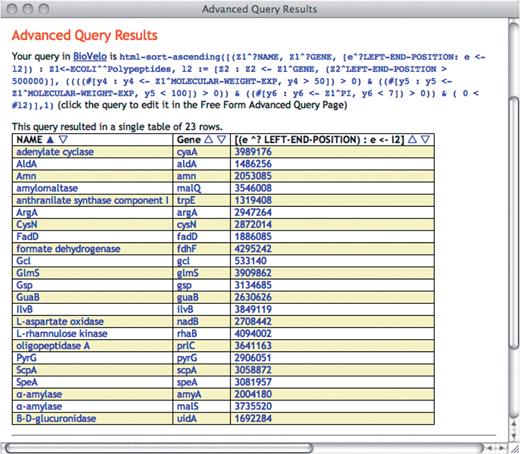
SAQP and schema documentation
Documentation is provided for several aspects of the SAQP and of the DB schema being queried. One form of documentation uses tooltips (small pop-up text windows that appear when the user hovers the mouse over a region of the screen). [For Internet Explorer (IE), we do not provide a tooltip mechanism since tooltips for selector elements do not work for IE (versions 6–8). Tooltips are provided for Firefox, Chrome and Safari.] Tooltips document the meaning of DB classes, DB attributes and for SAQP operators such as is equal to and is a substring of. Thus, this documentation describes both the workings of the SAQP and the schema of the DB being queried.
In addition, written instructions on how to use the SAQP and the FFAQP are available by clicking on the heading ‘Advanced Query Documentation’ at the top of the SAQP and the FFAQP.
To aid the reader in understanding subsequent examples, the following section presents a short introduction to the Pathway Tools schema that structures the data at BioCyc.org (and other Pathway Tools web servers) and describes how to learn more about the schema using the SAQP.
Pathway Tools schema
The schema of a DB is a plan of its structure. A schema describes the objects the DB contains and the relationships between them. Users face a significant barrier if they are forced to learn the schema of a DB before being able to query it. The approach taken by our interface is to provide enough guidance and online documentation to make learning of the schema incremental. The user can concentrate on the parts of the schema relevant to the desired searches.
The Pathway Tools schema is ‘object oriented’—it is based on ‘classes’ that are subdivided into ‘subclasses’. A class is a template based on a set of ‘attributes’, shared by all objects in this class. For example, the class Genes has the attributes product, and left-end-position (i.e. its left-end nucleotide coordinate in the genome), among others. (In this article, class names are capitalized whereas attribute names are not.) A subclass of a class inherits its attributes: the subclass is a child of the parent class. Typically, the subclass has more attributes, which makes its objects more specific than proper objects of the parent class.
Each attribute has a datatype that constrains the possible values of that attribute, such as number, string, Boolean, an object class or union of these. For example, the values of the product attribute of the Genes class must be objects within the classes Proteins or RNAs. Attributes also have a multiplicity, which can be ‘single’ or ‘list’. The attribute must have one value in the case of single, whereas it has zero or several values in the case of a list.
For example, the class Proteins has the attribute gene of type List of Genes. That is, this attribute has a value made of a list of one or more objects that represent genes (the list of genes that produce the protein). The attribute may have no values in the case where it is unknown what gene produces this protein. Although in the vast majority of proteins this attribute will have a single value, multiple values are used in the case where multiple genes within the genome all code for a protein having exactly the same amino acid sequence. The attribute dna-footprint-size, again of class Proteins, has type Integer. It is the number of nucleotides of the protein.
Most objects have one or more attributes that refer to other objects; we call such attributes ‘relations’. For example, the objects of class Reactions have the attributes left and right that refer to compound objects. They are, respectively, the reactants and products of the reaction. In general, these are lists of objects since several compounds may be on the right and left of the equation. The class Reactions also has the attribute in-pathway. It refers to the list of metabolic pathways in which the reaction is a part.
The relation component-of is used in several areas of the schema to define the part-of relationship. For example, the component-of relation defines the relationship between a protein monomer and a protein complex of which it is a part, and it defines the relationship between a gene and the chromosome on which it resides.
Schema exploration with the SAQP
Let us analyze how the SAQP can help the user discover relevant knowledge of the schema based on the query in Figure 2.
The user beginning to write this query may want to query proteins, but be unsure which of the several protein-related classes in EcoCyc to query. The user browses through the contents of the class selector, and alternatively mouses over three different EcoCyc classes related to proteins: Proteins, Polypeptides and Protein-Complexes. Documentation on each class is presented in a tooltip as the user moves the mouse over it in the selector. The user determines that Polypeptides is the desired class, because of wanting to select monomers only, not multimers, which are included in the other two classes.
Next, the user begins formulating the conditions of the query, and notices that two molecular weight-related attributes are available: molecular-weight-exp and molecular-weight-seq. The tooltip that appears when the user selects each one reveals that the first attribute refers to experimentally determined molecular weight, whereas the second refers to molecular weight computed from sequence. In our example, we have selected only the former, but some users might choose to include both. The documentation also indicates the units of a given attribute, kilodaltons in the case of these attributes.
Complex SAQP queries
The query presented in ‘Simple SAQP queries’ section contained a conjunction of conditions applied to one DB class (Polypeptides). This section and the two that follow explore the creation of more complex SAQP queries with different logical operators, quantifiers and more than one DB.
Combining logical connectors
The query in Figure 2 contains three atomic conditions, all of which are connected using the and connector. But in general, conditions can be connected by combining any of the four connectors that the SAQP provides for the creation of complex conditions. These are and, or, ornot and andnot. They are used to form complex conditions based on several atomic conditions. The connectors or not and and not apply a logical ‘not’ on the atomic condition before applying the corresponding connectors or or and, respectively. Notice that a condition is specified from top to bottom since when a connector is selected a new atomic condition is inserted below the last one. For example, for a condition of the form a and b or c, the meaning is (a and b) or c—it is not a and (b or c), which in general has a different meaning. For this particular case, the second meaning can be specified by reversing the order of the atomic conditions, that is by writing from top to bottom c or b and a. This is the case since these connectors are commutative. In other cases, some other reordering combined with one or several logical reversal (i.e. not equal versus equal) would make an equivalent condition.
Quantifiers on attributes of basic types
A ‘quantifier’ is used to query an attribute that has a list of values. We separate the discussions of quantifiers that apply to attributes with basic types such as numbers and strings, versus quantifiers that apply to attributes whose types are lists of other DB objects, which are described in the next section.
Imagine that we want to add an additional restriction to the query in Figure 2, to select polypeptides whose citations contain the word ‘purified’. To do so, we would add a condition and then select the citations attribute (no figure provided because of space limitations). This attribute has a type list of strings, typically, one string per citation. The user can then select the operator at least one element of, and enter ‘purified’ in the text box. Note that the citations attribute is a list of strings that contain evidence codes and PubMed/Medline identifiers. (This is documented online in the tooltip: the user can read a description of the citations attribute.)
Other quantifiers available for multivalued attributes with basic types include at least one element of, exactly one element of, for no element of and the number of elements of (which allows queries based on the number of values of an attribute instead of the values themselves).
Quantifiers on relations
A relation is an attribute whose values are other objects. In a sense, the attribute creates a relationship among DB objects. Example relations within the Pathway Tools schema include the attributes product (which links a gene to its product protein or RNA) and reaction-list (which links a pathway to its component reactions).
Quantifiers on relations within the SAQP allow us to realize a significant advance in functionality within an interactive query form by supporting a join-like capability. For example, imagine that we want to extend the query in Figure 2 with an additional restriction that depends on the ‘gene encoding the polypeptide’, not on the polypeptide itself. This type of condition is not supported in most other query forms.
To do so, the user would add an and condition, and then select the gene attribute, which represents the gene encoding the polypeptide (see Figure 2). As in the previous section, the SAQP rearranges the order of this condition to enhance its readability. We then select the quantifier operator for some object … , meaning that we want to define a condition that applies to some of the genes in the gene attribute of this polypeptide (although in the majority of cases only one gene will be present).
At this point the SAQP adds a new indented query clause as shown in Figure 2, to allow a condition to be defined on the gene. We have specified a constraint that its nucleotide coordinate must lie in the first 500 kb of the genome. Since several attributes and logical connectors can be specified in this new clause, forming a complex condition by itself, the web interface draws a box around this condition and introduces it with the we have keyword. A new unique variable, named Z2, is also introduced. This variable represents every value of the gene attribute. Variables are described further in the next section.
Additional quantifier operators applicable to multivalued relations are for all objects…, for one object … andfor no objects….
The quantifiers applicable to lists of basic values versus those applicable to objects are named differently: the word ‘element’ is replaced by ‘object’.
The use of quantifiers, with the exception of the number of elements of, generates a subquery in the underlying query language BioVelo (but is transparent to the user).
Quantifiers can be nested to any depth. That is, any of the atomic conditions under a quantifier can refer to an attribute that itself is a list on which a quantifier can be applied, and so on. Therefore, a long list of nested relations can be specified.
Queries with several components
Simple SAQP queries’ and ‘Complex SAQP queries’ sections presented examples based on only one search component. This restricted the complexity of the searches. For example, there were no searches in two or more DBs. Here, we show that adding a search component can provide more flexibility.
In the SAQP interface, a component can be added by clicking the button labeled ‘Select an operation to add an additional search component’. The addition of a component always introduces a new variable.
For two components, the second component does a search for each object found in the previous one; for three components, the third component does a search for each pair of objects from the previous two components; and so on. That is, the components do a search on the ‘Cartesian product’ of each list.
Figure 4 presents a query containing three search components: the first one in the BioCyc Escherichia coli DB, the second one in the DB for Helicobacter pylori strain 26695 and the third in the DB for H.pylori strain HPGA1. Notice that the button ‘Select an operation to add an additional search component’ still exists between the components. This provides the possibility to insert another search component between any already existing search components, not only at the end.
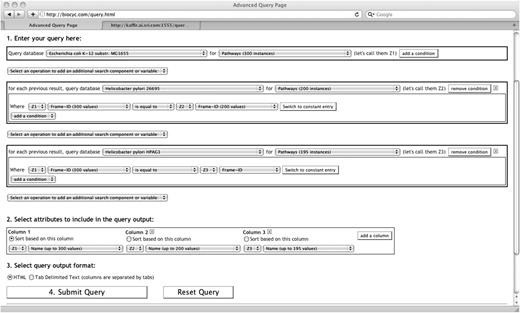
Three search components are used in this query across three DBs. This query searches for all pathways of Escherichia coli that also exist in two other DBs, Helicobacter pylori for strains 26995 and HPAG1.
This search uses the attribute Frame-ID to identify common pathways between databases. In general, the frame-id values are assumed unique for one database. Furthermore, a web server may offer, for some classes, to uniquely identify objects based on frame-ids across all its databases. In our case, if two pathways have the same frame-id value, regardless of the database, then this frame-id refers to the same pathway. Inversely, two different frame-ids are assumed to refer to two different pathways.
The query of Figure 4 is efficient, even though it has three search components, since the second search component keeps only the pathways that also exist, by frame-id, in Helicobacter 26695. It turns out that there are around 100 of these. The third search component, although searching through all the pathways of Helicobacter HPAG1, will do so for this small number of cases. Notice that the first component uses variable Z1 to identify every pathway of E.coli, that variable Z2 is used to identify every pathway of H.pylori 26695, and variable Z3 is used to identify every pathway of H.pylori HPAG1. These variables are introduced automatically by the interface. They are used in the conditions to form correlated atomic conditions. They are also used in the output specification to select appropriate attributes for the result. The following section presents more details about variables in the SAQP interface.
This particular example uses the Frame-ID attribute to compare objects. It is possible to compare (i.e. join) any database object attribute with any other database object attribute, as long as their type matches.
Variables
Variables are introduced automatically by the web interface. They make it possible to formulate precise conditions between objects in conditions. Without them, it is not possible to precisely refer to different objects across search components and conditions based on quantifiers.
When several search components or at least one quantifier are used in a query, explicit variables are introduced—they are always named Zn where n is an integer. Three variables, Z1, Z2 and Z3, are visible in various places in Figure 4. They cannot be explicitly renamed, created or removed by the user. The interface introduces and removes them as needed. Each one has a type (e.g. gene) since they represent objects from a class. The interface maintains the coherency of the list of attributes and operations that can be applied to each variable according to its type.
Each search component has at least one variable associated with it. This variable refers to the objects of the top (i.e. first) class selector. When only one search component exists, no explicit (i.e. visible) variable is associated with it—but it exists and it is called Z1. Therefore, when it becomes necessary to introduce a second variable, two become visible: the newly created one, typically called Z2, and the implicit Z1.
Each variable has a ‘scope’. The scope of a variable is the extent, in the query, where it can be used. The user does not have to worry about the scope of variables—that is, it is not possible from the interface to refer to a variable outside its scope. When a selector is provided to select a variable, at a specific location in the query, the list of choices contains the variables that are in the scope at this location—nothing more or less. Moreover, when a variable is selected, the list of attributes associated with this variable, typically provided as a selector, reflects the type of the variable selected. This greatly reduces the possibility of error compared with typical query languages like SQL.
The variable introduced when a new search component is created has a large scope: it can be referenced in all components below it and in the output specification and in all conditions of this search component. This means that any condition of a search component can refer to all variables above this search component including its own variable. A variable introduced when a quantifier (i.e. subquery) is specified has a scope to the condition under this quantifier and in the output specification. This latter case is very useful: it enables the user to output any attribute of the resulting objects of subqueries.
The right argument of any atomic condition can refer to either a variable or a constant. Using a variable makes it possible to create conditions between the current object of a search component and any other object in the previous search components. Similarly, in a condition based on a quantifier applied to the elements of a list, a variable is available to refer to the elements of the list. Any subcondition under the quantifier, which might also be based on a quantifier, can be based on this variable.
Implementation
The SAQP is implemented in about 3000 lines of JavaScript code. It has been tested on four browsers: Internet Explorer 6, 7 and 8, Firefox 3.2, Safari 4.0.3 and Chrome 4. Much of the complexity of the code results from the need to maintain consistency within the query. For example, selectable operators are type consistent with the attributes selected; selectable variables are in the scope of the expression being created; lists of selectable attributes are consistent with the type of variables; right operands make sense with the left attribute selected in an atomic condition; variables have unique names.
The translation of an SAQP query to BioVelo is relatively straightforward. It consists of a translation where the values of the SAQP selectors are translated into BioVelo operators. For example, the translation inserts the right BioVelo operators (e.g. ^ ^, ^, instring) in the right places, generates pairs of well-balanced parentheses for complex conditions and converts some logical connectors to two BioVelo logical connectors. The overall structure of the SAQP query already gives the structure of the BioVelo query.
The quantifiers within the SAQP create BioVelo subqueries. Each quantifier generates one or two BioVelo syntactic forms. They are always translated in the form of atomic conditions that use the BioVelo length operator # to count the number of elements of a subquery, then to be compared (e.g. ,< >, = ) with the constant value 0 or 1 depending on the quantifier.
The SAQP output section generates directly the head of a BioVelo query. It is always a tuple of the form where every zi is a variable and every ai is an attribute. The BioVelo html-sort-ascending function is always applied to the whole query based on the user-selected column.
As the user builds an SAQP query, no interaction occurs with the web server. All verifications are done in the user's web browser on the client machine. This approach greatly improves the speed of interaction, and is achieved by sending the entire schema to the client machine (as a JavaScript data structure), with all its documentation. The schema is necessary to create the list of attributes in selectors, do type checking and so on. Before the web server transmits the schema to the web browser, it queries the database, thus making the SAQP applicable to any database DB, not just Pathway Tools DBs.
Limitations
BioVelo is the underlying query language used by the SAQP. That is, the SAQP interface maps every query to BioVelo, but the following features provided by BioVelo are not accessible from SAQP.
In BioVelo, the head of a query specifies the output result. It can be any BioVelo expression. In the SAQP, the head of the query is always a tuple where each of its elements is a reference to an attribute. For example, this limits the result to a table where each element cannot be a subquery.
Arithmetic operations cannot be embedded within conditions. Such embedding can be useful, for example, when calculating the length of a gene, or verifying if a gene overlaps another gene by a few nucleotides. Also, basic statistics (e.g. averages) cannot be performed within the SAQP but can be done in BioVelo.
In BioVelo, subqueries can be inserted almost anywhere as they are expressions. In the SAQP, subqueries occur only when quantifiers are used.
A more advanced schema class browser would facilitate browsing of the classes within the Pathway Tools schema. Currently, only a subset of the classes is accessible to limit the complexity of the SAQP.
Related work
In comparing other web interfaces to the SAQP, we have ordered them according to their similarity to the SAQP, starting with the least similar.
Query-by-Example
Query-by-Example (QBE), developed by M. Zloof at IBM in the 1970s (6), is a graphical interface to query and modify relational DBs based on a table description. Using a table similar to the relation to query, a user specifies constant values or variables, along with some operators in some of the columns. The table description becomes ‘an example’ of the records to retrieve. Special ‘condition’ columns can specify relations between columns based on the variables. Several tables can be specified for several relations to do join operations. Such a description can be easily translated to SQL. Microsoft Access is a commercial product that offers a QBE-like facility. QBE, in this strict sense, is still problematic to query nonrelational biological DBs such as BioCyc.
Our SAQP includes a compact QBE interface for querying (not modifying) DBs. In QBE, a query is formed by specifying values, variables and conditions in a table of the same form as the relation being queried. This is visually appealing since all the attributes (i.e. fields, columns) of the relation are visible while creating the specification. On the other hand, if the relation has a large number of attributes, the visual aspect can become overwhelming. The remedy is to allow the user to remove and reorder the attributes as needed. Among other things, the SAQP allows this: once a DB and class (a relation) are chosen, a where clause can be added that has a selector of possible attributes. Any number of attributes can be added by using the and logical connector. QBE allows several relations to be queried in the same query, with possible sharing of variables to interlace the conditions on which to filter in the needed rows. The mechanism of several components in SAQP offers a more general mechanism.
Previous advanced query form at biocyc.org
The previous advanced query form at BioCyc.org, which has been superseded by the interface presented in this article, had a fixed structure. A search would be done by selecting a DB (e.g. E.coli), a class of objects (e.g. compounds), a connective (e.g. and), one or several slots (i.e. attributes of the class), constant values, and various constraints (e.g., greater than) between these slots and values. Depending on the chosen operator, such a search returns a list of objects satisfying all (e.g. and) or some (e.g. or) of the constraints on the slots.
It allowed only a single logical operator and limited the number of attributes. The main limitation is that it was not possible for queries to span multiple object types. Also, no subqueries were allowed.
EuPathDB
The EuPathDB (7) web site contains a page called ‘All Queries and Tools’ that provides a large number of possible queries to the user. Compared to the SAQP, the approach is quite different: EuPathDB uses a large number of predefined queries where each query allows a few attributes to be constrained. There are no facilities to create more precise queries based on several object types or with any number of user-defined constraints. For example, there is a set of queries called ‘Identify Genes by’ with a subsection called ‘Putative Function’. That subsection links to six different query pages: ‘Go Term’, ‘EC Number’, ‘Metabolic Pathway’, ‘Y2H Interaction’, ‘Predicted Interaction’, and ‘Phenotype’. Each query page is tailored to a specific search; some searches require the user to select one or several databases and enter one constraint, e.g. a GO term.
FlyBase, TAIR and Entrez
The majority of biological DBs allow user queries to one class of objects at a time, but do not allow queries that span multiple classes. This is true of the FlyBase QueryBuilder (see flybase.org/cgi-bin/qbgui.fr.html) and TAIR searches.
Entrez is subject to similar limitations, although the Entrez web interface offers a cross-database search mechanism. By entering a single string, several DBs are searched for this string. Since the DB searched contains different types of object, several types of object are returned. But the flexibility of the search is limited since multiple contraints cannot be specified, nor can interrelations between object types be specified.
BioMart
BioMart (8) is an open source data management system used by several web sites: WormBase, Rat Genome Database, UniProt, Reactome, Galaxy and others. For example, the WormBase web site, at wormbase.org, covers one organism, Caenorhabditis elegans and offers a query page based on BioMart. It has a few databases since a few versions of the wormbase are available.
The web query page provided by BioMart has two main panes: a left narrow one and a right wider one. The left pane acts as a menu from which the right pane can be changed to specify the data set to search, the output attributes and the filters to apply. It also summarizes the current query. A second query can be formulated below the first one.
The underlying query language is Perl using the BioMart libraries. The user can get the Perl code (as text) used for a specific query or an XML file representing the selections made for the query.
Biomart has several shortcomings compared with the SAQP. It is not possible to do join queries specified by the user. Only one logical operator, and, is available between the filter elements; the logical or operator is not available. BioMart includes a form of ‘not’ but on an individual filter element only. More important, the user must use one of the predefined filter elements. The attributes always have a single value, lacking the rich list structure to create multiple subqueries based on list and set of elements and objects.
Biozon
Biozon (biozon.org) integrates several biological DBs and provides a web interface to query them (9). A query is created by first selecting an object type, entering some constraints for this type and then proceeding to another related object type if desired. That is, join operations between different types of object can be done. This is one of the few web interfaces available today that allows joins. However, the number of relations used in Biozon is limited to a handful, and appears to be hardwired into the system. Further, the Biozon interface does not provide the full set of operators provided by the SAQP that distinguish all possible relationships between objects (e.g. find a protein such that all its genes satisfy some condition, or some of its genes, or none of its genes). Also, the SAQP allows the user to see the entire query in one page.
FlyMine
FlyMine has a complex querying interface called ‘QueryBuilder’ accessible at (10). The first step in creating a query is selecting a class of objects, such as ‘Gene’ or ‘Protein’. The next Web page is then divided into left and right panes. The left pane shows a user-expandable tree of attributes for the selected class of objects. For each such attribute, the user can either ‘show’ or ‘constrain’ it, or ‘summary’ or ‘constrain’ it. The show command adds the selected attribute to the output of the query. The constrain command allows the user to filter the results of the query to objects with the selected attribute having some specific values. For example, for class Gene, the attribute length can be constrained to be smaller than 1000. The user would select the relational operator < and enter the value 1000 in a text box. Each such added constraint requires communication with the web server, which can be slow. It is not possible to refer to other attributes in the constraint, only a constant can be entered.
Compared to the SAQP, this interface has the following limitations: it is not possible to search more than one DB in the same query, there are no quantifier operators on lists of objects, only one search component is allowed with one class of objects, and the constraints are based on constant only with no possibility to refer to other attributes of the same object. This last capability is possible in the SAQP since variables are introduced for each new search component.
BioGuide
BioGuide (11,12) is a user–centric framework which captures user preferences and querying strategies, and allows various querying approaches. The framework consists of a high-level, semantic view of the scientific domain in a Entity graph (e.g. Gene, Disease, Protein), that is mapped to the data sources of interest in a Source-Entity graph (e.g. SwissProt, UniGene, TrEMBL). Users pose queries over the Entity graph and are given a set of ranked paths in the Source-Entity graph that take their preferences and querying strategies into account. BioGuide helps scientists choose suitable data sources, find complementary information in sources and deal with divergent data.
BioGuide offers a graphical interface based on oriented graphs to query DBs (e.g. Entrez). Two oriented graphs are initially displayed. On the left is the Entity graph, and on the right is the Source-Entity graph.
The implementation does not offer data filters based on complex conditions using logical operators (e.g. or) nor to search entities based on specific attributes. For example, it is not possible to search a gene based on the molecular weight of its product.
BioGuide differs greatly from the SAQP. The SAQP emphasizes precise queries where the user has access to the database schema, whereas BioGuide emphasizes to search alternative paths for complementary results without a direct access to the database schema. The BioGuide interface implementation is based on the Java Web Start technology that eases the installation of a Java application. In contrast, the SAQP requires no additional application besides a web browser.
SAQP evaluation
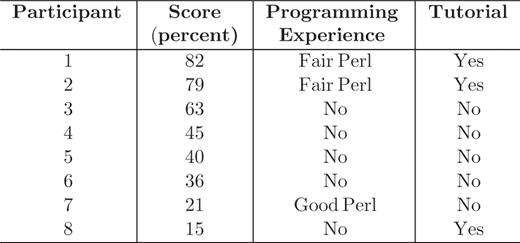
A user evaluation of the SAQP interface took place in May 2009 at SRI International in two different sessions. The two sessions differed in that the first cohort of five participants received no tutorial (nor any form of introduction) to the SAQP, whereas the second cohort of three participants watched a video tutorial of 10 min at the start of the session. They watched the same video tutorial that is available in conjunction with the SAQP through the BioCyc web site.
Common to both sessions were the following. The participants had never used the SAQP. Each participant received the same questionnaire of 10 precise queries to solve, using the SAQP, with an overall time limit of 1 h. The queries were written in English using standard biological terms. For example, one question was
Find all metabolic pathways in E.coli having more than 2 reactions and where none of the reactions has the reactant (i.e., the left side) “acetaldehyde” in it. How many such pathways have you found?
Each participant filled out a form answering basic questions about that person's own background. Five participants had no programming experience, two participants had fair Perl programming experience and one participant had good Perl programming experience.
The questionnaires were scored using the same criteria for all participants. The results are shown in Table 1.
The participant scores, their programming skills and whether they watch a 10-min video tutorial
 |
Analysis of evaluation results
The two highest scores were obtained by participants who had viewed the SAQP video tutorial and had Perl programming experience. The tutorial appears to have a positive impact although the lowest score was from the third participant, who viewed the tutorial. Experience in programming may help in using the SAQP, although this is not clear from this evaluation as the second-lowest score was from the participant who had the most programming experience. The participants who did not view the tutorial and had no programming experience had average scores.
The study participants exhibited a wide range of performance: 25% of subjects were able to answer most (75%) of the questions; 38% of subjects were able to answer more than half of the questions. Although these results say clearly that a high success rate is possible in a person's first encounter with the SAQP, we had hoped that more subjects would exhibit a high success rate. Clearly, not all subjects were readily able in one short period to solve the sample problems using the SAQP. Biologists who had some programming experience and were given a short tutorial were most capable of formulating simple and complex queries using the SAQP.
Since we are aware of no similar empirical evaluations for related query tools, it is unclear how other tools would compare in this evaluation. Such a study would indeed be an important research direction, but is beyond the scope of this work. Our work establishes a baseline for future comparisons.
The evaluation confirmed that understanding the schema is a key factor in using the SAQP. The problems within the questionnaire were formulated using a neutral vocabulary whose terminology sometimes varied significantly from the terminology (class and slot names) used in the schema. For example, one question asked the participants to find ‘all monomers in E.coli ‘ but the Pathway Tools schema uses the term ‘polypeptide’ rather than ‘monomer.’ Users reported that identifying appropriate schema terms was a significant challenge.
Results and conclusions
We presented the SAQP web interface for dynamic authoring of precise DB queries. SAQP queries combine a level of expressiveness and usability by nonprogrammers that is unrivaled among biological DBs. The SAQP is implemented as part of the Pathway Tools software, and translates its queries to the BioVelo query language for evaluation, although the syntax of BioVelo is hidden from the user.
The SAQP interface is available at BioCyc.org/query.shtml for more than 500 PGDBs. Development of the SAQP was informed by feedback from several EcoCyc and MetaCyc biologist curators. During 2009, an average of 855 SAQP queries per month were received at BioCyc.org, for 1252 unique visitors, illustrating that the SAQP is in active use by BioCyc users.
SAQP queries consist of a set of search components and a description of how the query output should be formatted. Each search component searches one DB; a query with more than one component can search multiple DBs. A search component specifies a set of conditions on a DB class to be searched. In addition, conditions can apply to other objects of other DB classes that are connected via relation attributes to the initial class. These conditions can be nested arbitrarily. Variables that appear within SAQP expressions allow the user to relate or compare terms that appear in spatially separated parts of a query.
One of the difficulties users face in querying a complex biological DB, such as those in the BioCyc collection, is understanding the DB schema. We cannot expect a user to learn or know this schema before creating a query. The SAQP interface provides basic tools to learn the parts of the schema required for a query. The tools are based on inline tooltips available for every schema attribute and class, which are generated within the SAQP by the web server. The SAQP also performs type checking to help the user make legal selections of variables, attributes and operators.
Funding
National Institutes of Health (grant GM75742). Funding for open access charge: GM75742.
Conflict of interest. None declared.
Acknowledgement
Sarah Cohen-Boulakia kindly provided several written clarifications about BioGuide. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
References
Appendix 1
The BioVelo language
BioVelo is the DB query language into which SAQP queries are translated. That is, every request formulated by the user in the SAQP interface is translated into BioVelo before being sent to the server for evaluation. When a user submits a query, the translated BioVelo query is displayed alongside the returned result (see Figure 3 for an example). A list of example BioVelo queries is provided at BioCyc.org/query.shtml. The specification of BioVelo is at BioCyc.org/BioVeloLanguage.shtml.
BioVelo can be applied to object-oriented DBs. It has set, list and tuple as nonatomic data type. Its syntax and semantics are based on list comprehension and it has an efficient implementation in Lisp described in (13).
For example, to retrieve all proteins from E.coli:
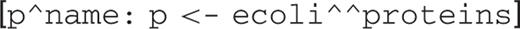
The head of the query is p^name. The operator ^ references an attribute. In the case of p^name, it references the attribute name of variable p. The p <- ecoli^^proteins is a generator. The name of the variable p is chosen by the user, but the name of the DB ecoli is fixed by the server, and the name of the class proteins is fixed by the schema of the server. The dyadic operator ^^ references a list of those DB objects that are instances of the specified class. In the case of ecoli^^proteins, it references all proteins from the E.coli DB. This generator means: for each object of the class proteins in the DB E.coli, bind variable p to it. Since there is nothing after the generator, the head is executed, which simply retrieves the name of the protein. The list is built sequentially. This is essentially the semantics of list comprehension: the generator creates a ‘loop’, and the head expression creates the result for each value generated by this loop.
This example had only one generator, but constraints, subqueries and more generators can be added. For example, the query shown in Figure 2 is translated into

This last query has one main generator, with variable z1, four subqueries, one of which is bound to variable L2, and four atomic conditions forming one conjunctive condition. The head of the expression is a tuple of three expressions: the name of the polypeptide, the gene(s) producing this polypeptide and the left-end position(s) of the gene(s) on the genome. This tuple forms a table of three columns. The operator # gives the length of a list. The operator ^? is similar to ^ but it generates a URL link for HTML display.
BioVelo offers basic arithmetic operations, set and list operations, indexing and so on. Queries can be embedded. In fact, a query is an expression and can be specified anywhere an expression can be specified, as long as its type is compatible with the operation done on it. Some aspects of BioVelo are not accessible from the SAQP interface. For example, arithmetic operations available from BioVelo cannot be done from the SAQP.
The FFAQP gives full access to the BioVelo language—this form is accessible via the SAQP at (14).

{kind=link}
{kind=link}
{kind=link}
{kind=link}