





Abstract
The use of high-throughput RNA sequencing technology (RNA-seq) allows whole transcriptome analysis, providing an unbiased and unabridged view of alternative transcript expression. Coupling splicing variant-specific expression with its functional inference is still an open and difficult issue for which we created the DataBase of Alternative Transcripts Expression (DBATE), a web-based repository storing expression values and functional annotation of alternative splicing variants. We processed 13 large RNA-seq panels from human healthy tissues and in disease conditions, reporting expression levels and functional annotations gathered and integrated from different sources for each splicing variant, using a variant-specific annotation transfer pipeline. The possibility to perform complex queries by cross-referencing different functional annotations permits the retrieval of desired subsets of splicing variant expression values that can be visualized in several ways, from simple to more informative. DBATE is intended as a novel tool to help appreciate how, and possibly why, the transcriptome expression is shaped.
Database URL:http://bioinformatica.uniroma2.it/DBATE/.
Introduction
Alternative splicing (AS) permits the synthesis of multiple transcript variants from a single gene, thus increasing the diversity of RNAs and proteins encoded by a genome (1, 2). The number of known splicing variants in the human transcriptome stored in Ensembl is growing at a dramatic pace. Through the use of recent high-throughput RNA sequencing technologies (RNA-seq), it has been demonstrated that ∼95% of multi-exon genes undergo AS in panels of human tissues (3), shaping the expressed transcriptome in various ways (4) and generating an exceedingly complex repertoire of mRNAs (5). Splicing variant expression deconvolution algorithms, such as Cufflinks (6), IsoEM (7), Scripture (8), RSEM (9) and SpliceSeq (10), allow the reliable (as validated in many instances by RT-PCR) quantitative estimation of the transcription of individual splicing variants of a gene from RNA-seq data. Yet, the functional interpretation of such expression data, or the change of splicing variant-specific expression patterns in different tissues or conditions, is still overly difficult. In the simplest cases, splicing promotes the inclusion or removal of specific exons corresponding to whole-protein domains to which a specific function can be assigned, but often splicing patterns are much more complex and the effect of splicing on the protein product function(s) is much more elusive. A number of indications suggest that in a considerable fraction of cases, splicing can radically change the protein product function and/or fold (11–14), and a non-negligible amount of splicing variants shows structural inconsistencies (e.g. low degrees of residue packing in the protein core or large fractions of hydrophobic residues exposed to the solvent), and lack of known functional regions. As a consequence, despite the large amount of data available about AS variants and protein functional annotations, there are no resources dedicated to the integrated retrieval of such information.
A number of databases offer storage or download of next-generation sequencing data (15, 16), but the splicing variant-level expression analysis is still unfriendly for the biomedical researchers, given the exceedingly large amount of data to be processed, the computational power required and the nature of the analysis algorithms that are usually intended for the computational biologist. A small number of recent databases storing RNA-seq expression data only provide gene-level expression values (e.g. 17). Splicing variant-level annotations are starting to be available in databases, such as Uniprot, but they can be of difficult interpretation without a reference context, and are still largely incomplete. SpliceSeq (10) provides a user-friendly interactive graphic environment, integrated with isoform-specific functional annotations from Uniprot, but splicing variant-level expression estimation must be run by the user. Many databases exist that collect AS variants (18–21), but they do not tackle variant functional annotation. There are no general tools that can be used to infer whether a given variant is actually translated, and its eventual protein product stable and containing functional regions and residues. Various resources have been developed for the analysis of specific effects of AS, for example ProSAS (22) for the analysis of the changes introduced by AS on protein structures, or AS-ALPS and AS-EAST (23, 24) for the analysis of the effect of AS on protein–protein interfaces and other structure-based functional assignments. A web server, MAISTAS (25), provides a framework to test the structural consistency of a splicing variant, but has a limited range of application because it requires a high sequence identity between the variant under analysis and a template with known 3D structure. As a consequence, the integration of transcript-level RNA-seq expression and their functional characterization must be currently approached by combining different tools and cross-referencing heterogeneous databases and data types.
We aim at filling this void with the DataBase of Alternative Transcripts Expression (DBATE). We processed 13 large public RNA-seq panels from human healthy tissues and in disease conditions. For each splicing variant in each sample, we report the estimated transcript expression and its functional annotations, extracted and integrated from different sources: Ensembl (26), Pfam (27), Uniprot/Swiss-Prot (28), GO (29) and mentha (Calderone & Cesareni, in press; http://mentha.uniroma2.it/). The user can access splicing variant expression levels of the genes or transcripts of interest, compare them among different samples and perform more complex queries by cross-referencing the available annotations. The interface is designed to facilitate the data retrieval, available in five different formats: HTML tables, Excel spreadsheets, plain text tab-separated files, barplots and heatmaps.
DBATE content
DBATE provides the expression level for each human transcript annotated in Ensembl (release 67) estimated in 13 different panels of human tissues/cell lines available in the Gene Expression Omnibus (GEO) (30), enriched with functional annotation. These panels have been chosen to cover the largest number of samples from human healthy tissues, organs or cell lines; for seven of them, normal and tumoral condition is provided. Each sample in each panel was processed independently, and we provide tools for the comparison of any given set of samples that the user can select as desired. The list of available data sets is provided in Table 1 reporting, for each data set, its GEO identifier, the samples it contains, the total number of reads, the sequencing technology used, a description of the data set content and the literature reference (when available).
Data sets included in DBATE
| GEO GSE identifier | Samples | Number of reads (×106) | Read length (bp) | Description | Reference |
|---|---|---|---|---|---|
| GSE12946a | Adipose, brain, breast, colon, heart, liver, lymph node, skeletal muscle, testes, BT474, HME, MB435, MCF-7, T47D | 224 | 32e | The Wang data set, from which we selected 14 samples, 9 in normal condition and 5 in tumoral condition | 31 |
| GSE17274b | Three female (HSF1, HSF2, HSF3) and three male liver samples (HSM1, HSM2, HSM3) | 72 | 35e | Sex-specific gene expression in liver in three males and three females | 32 |
| GSE29119b | Breast cancer (HCC1954) and normal breast cells (HMEC) | 97 | 36e | Gene expression analysis of breast cancer | 33 |
| GSE29155a | Prostate epithelial (PrEC) and prostate adenocarcinoma (LNCaP) cell lines | 9 | 36e,40f | Transcription profiling of human prostate epithelial and adenocarcinoma cell lines | 34 |
| GSE29580b | Normal and tumor samples from two colorectal cancer patients | 40 | 36e | Whole transcriptome sequencing of colorectal cancer | NA |
| GSE29968b | Matched esophageal squamous cells from three carcinoma patients | 118 | 38e | Transcriptome analysis of human esophageal squamous cell carcinoma in three pairs of matched patient-derived tumor samples and their adjacent non-tumorous tissues | 35 |
| GSE30611c | Adipose, adrenal, brain, breast, colon, heart, kidney, liver, lung, lymph node, ovary, prostate, skeletal muscle, testes, thyroid, white blood cells | 2000 | 50f | The Illumina BodyMap 2.0 Project, comprising transcription profiling of individual and mixtures of 16 human tissues | NA |
| GSE30772c | Mitochondrion, mitoplasm | 45 | 35e | Examination of the mitochondiral transcriptome | 36 |
| GSE32689b | Pooled oocytes, pooled sister polar bodies, single oocyte, single sister polar body | 120 | 42e | Transcriptome of the human polar body, providing four conditions: pooled oocytes and their sister polar bodies and a single oocyte and its sister polar body | 37 |
| GSE33328d | Peripheral brain tissue, tumor brain tissue | 49 | 75e | Transcriptomic profiling of a glioblastoma multiforme patient with control peripheral brain tissue | 38 |
| GSE37769c | THP1 cells | 287 | 100e | Expression analysis of the THP1 (human monocytic leukemia) cell line | 39 |
| GSE38685b | Prostate epithelial (PrEC) and prostate adenocarcinoma (LNCaP) cell lines | 35 | 75e | Transcription profiling of human prostate epithelial and adenocarcinoma cell lines | 40 |
| GSE43925b | THP1 high glucose, THP1 normal glucose | 60 | 42e | Expression analysis of human THP-1 monocytes in normal conditions and treated with high glucose | 41 |
aPlatform: Genome Analyzer
bPlatform: Genome Analyzer IIx
cPlatform: HiSeq 2000
dPlatform: Genome Analyzer II
eSingle end reads
fPaired end reads
The current DBATE release includes 13 data sets retrieved from the Gene Expression Omnibus (GEO). The Table table reports for each data set its GEO GSE identifier, the samples it contains, the total number of reads (expressed in million reads), the read length, a brief description of the data set content, and the literature reference when available (NA indicates that the data were deposited in GEO but the study is still unpublished). Superscripts indicate the sequencing technology employed used (either GA, GAII, GAIIx, or HiSeq 2000), and whether the reads were sequenced as single or paired ends.
RNA-seq analysis
All the data sets have been checked for read quality using FASTQC (v0.10.1 at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). We trimmed read ends if base quality scores were <20. At the end of this process, if the total length of the read is <10 bases, we discarded the read.
We used the Tuxedo Suite, comprising Bowtie (v0.12.7) (42), TopHat (v1.4.1) (43) and Cufflinks (v1.3.0) (6), to align the sequence reads produced in each experiment to the reference human genome hg19 seeking only unique genome matches with up to two mismatches (Bowtie), to identify splicing junctions (TopHat), and to evaluate the normalized expression of individual splicing variants (Cufflinks) reported in Fragments Per Kilobase of transcript per Million mapped reads (FPKM). Cufflinks is the de facto standard algorithm for the isoform deconvolution problem, and it is widely used (44). More recent algorithms are available (45–48), but in absence of appropriate benchmarks for this kind of algorithms we deemed more appropriate to select the most popular tool. We actually tested two additional splicing variant expression estimation algorithms, IsoEM (7) and Scripture (8), on the Wang data set (see above), finding a high correlation with the Cufflinks expression estimates (Pearson correlation coefficient >0.8 for both algorithms).
Functional annotation
Various sources of functional information have been integrated in DBATE, gathering functional annotations from the Ensembl, Uniprot/Swiss-Prot, Pfam, Gene Ontology and mentha databases. With the exception of protein interaction information from mentha (described later in the article), all annotations are mapped to individual splicing variants. Each feature can be used to filter the input query, selecting only those genes having at least one splicing variant carrying the desired annotation or set of annotations. It should be noted that, although the annotation transfer pipeline is intended for protein-coding transcripts, DBATE also stores expression levels for non-coding RNAs, which are assuming a central role in many cellular processes (49, 50).
We extracted from the Ensembl database (release 67) the definition (i.e. the exon composition) of each transcript linked to its gene (ENSG), transcript (ENST), associated protein (ENSP) identifiers and its genomic coordinates, defined by chromosome, absolute start–end on the human genome and strand. In total, DBATE stores 22 403 protein-coding Ensembl genes and their associated 100 357 transcripts, for which 94 737 (94.4%) encode for a different protein sequence (the remaining variants differ only in the untranslated regions). Additionally, DBATE stores 96 136 non-coding transcripts, some of which (52%) are non-coding isoforms of protein-coding genes, whereas the remaining 48% are products of genes for which no transcript is translated that might encode for functional RNAs, i.e. RNAs that are not translated because they have no ORF, but exert their function as RNA molecules (51, 52). A rapidly increasing interest in these non-coding RNA (ncRNAs) genes motivated their inclusion in DBATE, even if obviously in these cases we cannot apply our protein annotation transfer pipeline because these genes do not encode for proteins.
We collected all the human entries of the Uniprot/Swiss-Prot database resulting in 20 231 different entries. For each entry, we gathered the Uniprot ID, Uniprot Primary Accession Number, sequence of the main splicing variant, protein existence evidence, features and post-translational modifications. All the information stored in Uniprot is related to the main splicing variant protein product. This splicing variant is usually selected as the longest one, or the more biologically relevant (e.g. more commonly expressed, or better characterized) or the first discovered one. We mapped 16 distinct Swiss-Prot features to Ensemble transcripts, selected as those that are more directly linked to the protein function. These features are TOPO_DOM, NP_BIND, REGION, BINDING, DISULFID, MOTIF, MOD_RES, DOMAIN, DNA_BIND, REPEAT, ZN_FING, LIPID, ACT_SITE, METAL, SITE and CA_BIND. A total of 36 728 Ensembl transcripts encode for proteins that are annotated with at least one Swiss-Prot feature. Additionally, we also mapped 19 post-translational modifications provided by Swiss-Prot entries to Ensembl transcripts: acetylation, ADP-ribosylation, amidation, disulfide bond, gamma-carboxyglutamic acid, glutathionylation, glycation, glycoprotein, hydroxylation, iodination, lipoprotein, methylation, myristate, nitration, oxidation, phosphoprotein, S-nitrosylation, sulfation and Ubl conjugation. A total of 27 867 Ensembl transcripts encode for proteins that are annotated with at least one post-translational modification. The annotation transfer pipeline is based on the mapping between each protein amino acid and its corresponding genomic codon, identified as chromosome, strand and a triplet of genomic positions. We aligned each human Uniprot sequence using the Needleman–Wunsch algorithm with each Ensembl transcript sequence to find correspondence between the Uniprot sequence and all splicing variants of its encoding gene. Using the genomic coordinates of each transcript exon and the alignment between the Uniprot sequence and the most similar transcript (discarding all cases where there is no clear correspondence with any Ensembl transcript), we mapped the genomic location of the codons encoding for each annotated amino acid residue and verified the presence of each codon in the splicing variants of that gene, obtaining an estimate of how many annotated residues are present in each known transcript.
This procedure permits to map each annotation at the amino acid residue level from the main splicing variant (as identified in Uniprot) to each other splicing variant. Each splicing variant of a gene may encode only for a subset of the residues associated with an annotation in the main splicing variant; therefore, we defined the annotation coverage as the fraction of annotated amino acids found in a given splicing variant with respect to the total number of annotated residues in the main Uniprot splicing variant. Coverage varies between 100 (all annotated residues included) and 0 (the functional feature is completely removed by splicing events). For example, in the case of a splicing variant containing 5 annotated residues out of 10 annotated residues in the main splicing variant, the coverage of the annotation on this splicing variant is 50%. Obviously, even if an annotation is found on a transcript with high or complete coverage, that splicing variant is not necessarily able to perform that function because we cannot estimate if and how much the transcript is translated, and also because the function might depend on a specific local or global folding, or on the presence of disjoined regulatory regions, which would be extremely difficult to infer. Yet, if the annotated residues are not encoded by a splicing variant, that variant cannot perform that function, regardless of its translation, folding and regulation. Hence, we provide a transcript-level transfer of functional amino acids, which can be a useful starting point for a more detailed functional characterization.
Protein-domain composition was retrieved from the Pfam database and mapped to individual splicing variants using the server edition of PfamScan: 70 298 transcripts encode for a protein product that has at least one Pfam domain.
Finally, 72 916 transcripts are annotated with at least one Gene Ontology (GO) term. In total, 51 015 transcripts are associated with at least one GO term in the ‘biological process’ domain; 63 868 transcripts are associated with at least one term in the ‘molecular function’ domain; 51 634 transcripts are associated with at least one term in the ‘cellular component’ domain. Ensembl and Biomart recently started to associate GO terms with individual splicing variants. Although such annotations are largely incomplete, yet they provide in many cases variant-specific information. GO term annotations will be frequently updated over time.
The mentha interactome database collected protein–protein interactions (PPI) retrieved from five different PPI databases—IntAct (53), MINT (54), DIP (55), BioGRID (56) and MatrixDB (57)—with the aim of eliminating redundancy between these different sources. The main motivation of the mentha database is that currently available curated databases of protein interactions offer only a limited view of the interactome which can be expanded and made more consistent by their integration. We have combined this database with DBATE through a Java Applet to browse PPIs using the unique Uniprot ID associated to each transcript.
Querying DBATE
The DBATE database archives a wide variety of functional annotations. The user can access the splicing variant expression level of genes or transcripts of interest, or perform more complex queries using an advanced form through the use of cross-referenced annotations.
The DBATE user interface is intended to provide the choice of increasingly complex queries, in an intuitive fashion. In the simplest query, the user inputs a transcript ID, and retrieves pre-computed expression levels in FPKM units for all tissue samples in BodyMap (the default sample group), a simple TAB delimited plain-text file, a Microsoft Excel table file and an HTML table. All functional annotations of the input transcript are reported and organized in panels. When the user inputs a gene ID or a gene name, all its splicing variants are returned. Moreover, the database can be accessed via one or more GO terms or Pfam domain IDs; mixed queries are also allowed. Individual variants can be manually selected from the list of variants matching the query. In addition to the different tabular outputs and annotation panels, if the number of variants is higher than one (and lower than 100), DBATE also offers a heatmap grouping similar expression patterns across all selected samples.
Using the advanced options input form, the user can create more complex queries. First, specific samples from all RNA-seq data sets can be selected. Then, functional annotations can also be chosen to filter the input transcripts for only those matching the selected features. As a case study, we report the analysis of proteins containing the K Homology (KH) domain (Pfam ID: PF00013), a domain able to bind RNA promoting its degradation and that is involved in splicing regulation (58, 59). The KH domain has been found in some cases associated to repeated protein sequences such as the ankyrin repeat (60, 61), and its phosphorylation can modulate its binding ability (62). DBATE can be easily queried to retrieve all splicing variants that encode for protein products that contain the KH domain; then the query can be refined to retain only splicing variants encoding for phosphorylation sites and containing repeat units in their protein sequences, and for retrieving expression values in the desired pool of samples. Such a composite query that cross-links different types of information retrieves 10 transcripts belonging to 4 different genes: ANKRD17, KHSRP, HNRNPK and ANKHD1. Their expression patterns are reported in the heatmap in Figure 1, showing that splicing variants for these proteins can have different and tissue-specific expression. Interestingly, not all the ANKHD1 variants contain the KH domain. An ANKHD1 splicing variant lacking the KH domain, which has important roles in apoptosis, is reported in the literature (63). Retrieving the full list of which ANKHD1 variants contain the KH domain, and their expression patterns, is not a trivial task, but can be immediately obtained from DBATE by simply querying the ANKHD1 gene. DBATE reports that 10 out of 19 ANKHD1 splicing variants lack the KH domain, and their expression patterns can help in elucidating their cellular roles.
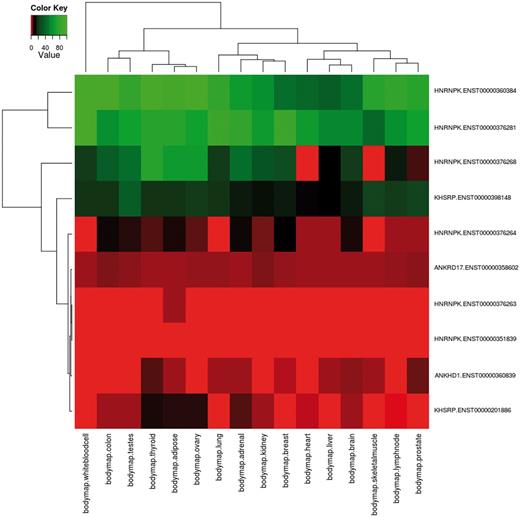
Example of combination of complex queries in DBATE. This heatmap reports expression values in the BodyMap panel of human tissues of splicing variants that encode for protein products containing the Pfam KH domain (PF00013), which are phosphorylated and contain repetitive units. The combination of this information can be easily obtained using the web interface of DBATE that returns in this case 10 different splicing variants that belong to genes ANKRD17, KHSRP, HNRNPK and ANKHD1. Their expression patterns show that splicing variants for these different proteins can have tissue-specific behaviors. The heatmap image is generated by an automated procedure using the statistical software R using the heatmap.2 function, and then loaded on the web interface as part of the results page. The color code of the heatmap ranges from red, lower FPKM values; to black, medium expression values; to green, higher expression values.
For each query, protein interaction data from the mentha browser can be visualized through a Java applet integrated in the results page. For each splicing variant derived from the query, a list of unique Uniprot primary accession numbers is used to interrogate mentha. For each query protein, all physically interacting proteins retrieved in mentha are reported as connected by an edge to the query proteins, and the expression FPKM in a selected tissue of each transcript is reported, for both the query proteins and their binding partners. Each network node is color-coded by the expression level of the dominant isoform, whereas clicking the single node reports all the different splicing variants with their expression values. The mentha browser additionally allows expansion and pruning of the network. The interaction network for the 4 KH domain-containing genes selected in the previous paragraph is reported in Figure 2, where they display close connectivity mediated by common binding partners.
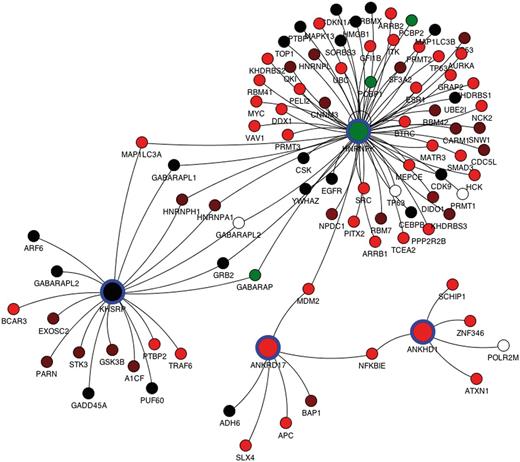
Protein interaction network for the ANKRD17, KHSRP, HNRNPK and ANKHD1 genes retrieved from the mentha database and plotted by the mentha browser applet. The mentha database stores manually curated PPIs from five different PPI databases and has been implemented in the DBATE web interface. These four genes have been selected from a complex query search on DBATE to obtain all the splicing variants that encode for protein products containing the KH (K Homology) domain and that are also phosphorylated and contain repeated units. The network includes all primary binding partners of the four genes. Nodes describe genes, and arcs join genes whose protein products are known to physically interact. Nodes corresponding to the query proteins are larger and highlighted with blue circles. Each node is colored according to the expression level of its most expressed splicing variant. Color ranges from red, lower FPKM values; to black, medium expression values; to green, higher expression values. White nodes describe genes for which no splicing variant is expressed in the selected tissue. Protein interaction networks generated by the mentha browser can also be manually expanded and pruned.
Finally, expression data from all data sets and transcripts can be downloaded as static tab-separated text files.
Data organization and web interface
DBATE has been implemented within the MySQL database management system version 5.1.17 on a Linux Xubuntu server machine. It contains expression levels for 196 494 splicing variants (57 659 genes) computed in 36 samples: 28 in the normal condition and 8 in the tumoral condition. For each splicing variant, information related to Ensembl, Uniprot/Swiss-Prot, Pfam and GO has been integrated as explained in paragraph ‘Functional Annotation’.
The web interface is implemented through python-CGI programming, HTML and JavaScript. All the graphs are generated through the statistical software R v.2.15.2 and loaded to the web interface through python-CGI. The entity–relationship schema of the database is included in the online DBATE help pages.
Conclusions
Next-generation sequencing technologies revolutionized the analysis of the transcriptome, providing a panoramic view of all the transcriptional activity in a given sample. Although such high-throughput experiments provide an enormous wealth of data, there are few tools to make order through them. DBATE, freely available at http://bioinformatica.uniroma2.it/DBATE/, provides an integrated resource that can be valuable for the functional inference of whole transcriptome expression analysis experiments by providing pre-computed expression levels and annotations that can be cumbersome to generate for the biomedical scientist.
A semi-automated pipeline was built to process and populate the database with additional data sets as they become available, and will be expanded to more annotation sources and sequencing technologies. The pipeline is based on initial steps of data retrieval, organization and quality checking, done manually by DBATE curators, followed by automated stages of expression estimates and annotations. We initially selected for inclusion in DBATE public data sets from a list retrieved from GEO, including 53 RNA-seq human panels obtained with Illumina technology (GA, GAII, GAIIx or HiSeq). All remaining unprocessed data sets are currently in a queue and will be progressively added. DBATE updates are planned each semester. Finally, DBATE will be expanded into a web server or web service-based tool for the annotations and characterization of user-submitted RNA-seq panel data.
Funding
Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG10298 to M.H.C.). Funding for open access charge: PRIN 2010 (prot. 20108XYHJS_006 to M.H.C.).
Conflict of interest. None declared.
References
Author notes
Present address: Valerio Bianchi, Center for Genomic Science of IIT@SEMM, Istituto Italiano di Tecnologia, via Adamello 16, Milan 20139, Italy.
Citation details: Bianchi,V., Colantoni,A., Calderone,A., et al. DBATE: database of alternative transcripts expression. (2013) Vol. 2013: article ID bat050; doi:10.1093/database/bat050.

{kind=link}
{kind=link}