





Abstract
High-quality manual annotation methods and practices need to be scaled to the increased rate of genomic data production. Curation based on gene families and gene networks is one approach that can significantly increase both curation efficiency and quality. The Sol Genomics Network (SGN; http://solgenomics.net) is a comparative genomics platform, with genetic, genomic and phenotypic information of the Solanaceae family and its closely related species that incorporates a community-based gene and phenotype curation system. In this article, we describe a manual curation system for gene families aimed at facilitating curation, querying and visualization of gene interaction patterns underlying complex biological processes, including an interface for efficiently capturing information from experiments with large data sets reported in the literature. Well-annotated multigene families are useful for further exploration of genome organization and gene evolution across species. As an example, we illustrate the system with the multigene transcription factor families, WRKY and Small Auxin Up-regulated RNA (SAUR), which both play important roles in responding to abiotic stresses in plants.
Database URL: http://solgenomics.net/
Introduction
Genome databases aim to reflect the state of the research in their species of interest, capturing information from experiments reporting single-gene studies as well as information from high-throughput studies involving thousands of genes. Database designs have to keep up with these developments and evolve to house the metadata from various novel technologies. The Sol Genomics Network (SGN; http://solgenomics.net) (1) is a clade-oriented database serving as a comparative genomics platform for Solanaceae species. The genome sequence of the tomato, Solanum lycopersicum ‘Heinz’, was published in early 2012 (2), presenting new curation challenges to capture the upsurge of new genomic information from the Solanaceae research community. The SGN database consists of a comprehensive community curation system (3) to which several new features have recently been added, such as tools to deal with gene family curation. Because the completed tomato genome sequence has become available, genome-wide profiles of specific multigene families are being studied rapidly. The results of these studies encompass complex sets of biological information that need to be curated into the database, and for which new curation tools and novel curation strategies need to be developed. While manual curation of single genes is a labor-intensive task, the manual curation of gene families can become quickly overwhelming if no specialized tools are used in the curation process. New specialized databases such as Ensembl Plants, PlantGDB (4), Phytozome (5) and PLAZA (6) provide powerful platforms to perform evolutionary analysis of plant gene families, and well-annotated gene family datasets carrying structural and functional annotations are a prerequisite for such analysis.
Members of a gene family are descendants of a common ancestor, generated as a result of duplication events and gene divergence (7, 8). Common structural features along with similar and diverging functional features of individual genes are characteristic of multigene families (7). Through their evolutionary history as well as structural and functional similarities, multigene families represent important reference points when they are accurately curated and enriched with annotations.
Here, we present a gene family–specific curation interface that addresses curational challenges posed by gene families, and also apply parts of this interface to the curation of gene networks.
Materials and methods
Literature curation
Solanaceae-related articles are downloaded from PubMed and prioritized by SGN curators. From the weekly/monthly downloads of hundreds of published articles, SGN curators manually identify articles that carry new information about Solanaceae genes/genomes. Articles that provide completely new information are given the highest priority to maximize the scarce curatorial resources available with the SGN team [<1 full-time equivalent (FTE)]. SGN curators capture as much information as possible about genes described in the publications, and then contact community curators, who are usually recruited from the authors of the publications (3). Such ‘locus editors’ have special database privileges that allow them to edit the basic information of the gene, such as name, symbol, function and description. Any user with ‘submitter’ privileges can annotate synonyms, alleles, phenotypes, publications, ontology annotations and create gene networks. The web interfaces used by community curators are the same as those used by in-house curators.
Structural loci and sequence curation
High-quality structural annotations are a prerequisite to support high-quality functional annotations. Most of the structural annotations available at SGN come from large sequencing projects, such as the tomato genome sequence hosted by SGN. This genome annotation was performed by an international consortium (International Tomato Annotation Group, ITAG), whose computational prediction of the tomato structural genes for the tomato genome assembly SL2.40 (annotation version ITAG2.3) yielded >34 000 gene models predicted with a combination of ab initio gene predictor tools and RNA-seq mapping data (2). The other Solanaceae reference genome is a double-monoploid accession of potato (Solanum tuberosum phureja) (9), which is mirrored on the SGN site, including its gene model annotation that was provided by Beijing Genomics Institute (BGI).
SGN also hosts two draft genomes, one of the tomato wild species, Solanum pimpinellifolium, a red-fruited tomato species and a close relative of the domesticated tomato. This genome was assembled de novo, with gene models based on the S. lycopersicum annotation. The other draft genome is of the allotetraploid tobacco species Nicotiana benthamiana, which is an important model for plant–microbe interactions (1). Annotations for genomes produced at SGN are usually annotated using the Maker pipeline (10), which combines ab initio prediction and experimental evidence.
Manual curation of structural annotations
The automated annotation of these genomes provide an important source of information for gene analysis; however, automated methods do not yet produce perfect results and sometimes require intervention using manual curation. Tools used for this purpose include GenomeView (11) and Apollo (12). Sequence alignments of gene family members can provide evidence for mis-annotations, as intron positions and other aspects of gene family sequences are often conserved. The SGN system provides a powerful sequence alignment module, which is linked to the gene family curation system.
Annotation of loci
The concept of a ‘locus’ in SGN is similar to that of the TAIR (13) and Gramene (14) locus, with a few notable differences. Functional loci are defined as genes with supporting experimental evidence, usually originating from the literature, but also from collections of monogenic Mendelian mutants (3). There are genetic loci from 12 Solanaceae organisms in SGN, but only two, tomato and potato, have a high-quality reference genome. The predicted structural genes, described in the ‘materials and methods’ section, were added to the locus database when these genomes became available, and merged with the existing genetic loci whenever applicable. This merging step is performed only when a curator decides there is adequate experimental evidence linking a previously described functional gene with a predicted gene model based on the genome sequence.
Each SGN locus carries information linking it to different modules of the database, capturing as much biological information as possible. In addition to a free text description, there are several mandatory fields, which include locus identifier, locus name, synonyms, gene activity and chromosomal position. Ontology-based annotations are made using an easy-to-use web interface that is based exclusively on pull-down menus and autocomplete forms, which helps ensure consistency and data integrity. Community annotation is open at the level of the functional gene (3); however, structural genes rely directly on the current standard genome annotation version, which are released at regular intervals. Quality control checks on community-curated entries are done by in-house curators. Apart from a detailed curation guidelines document, support to community curators is provided via the feedback option (sgn-feedback@solgenomics.net). In addition, SGN holds periodic curation jamborees for Solanaceae researchers. Usually these are held in connection with the yearly International SOL meetings for the locus editors.
Gene family curation
The first step in curating gene family information is to establish the nomenclature of the genes in the family. This step usually requires that the curator find the gene/protein accessions identifiers in the publication and map it to the sequence in the database, similar to single-gene curation. Names of gene families are recorded in addition to the individual gene name. Often, gene family members are given numerical suffixes at the end of the gene family name. For example, the Auxin Efflux Facilitator gene family members are denoted as SlPIN1 to SlPIN8. For each entry of the gene family, the gene name with the numerical suffix as well as the gene family name and synonyms are recorded so that queries of individual gene family members and entire gene families can be made from the search box. The tomato research community uses a variety of identifiers to report genes in their studies, ranging from SGN unigene IDs, genbank accessions (15), the genome gene model ID or chromosome coordinates. The gene model ID (‘Solyc id’) is the ideal accession, as it maps directly to the gene information in the database. After a gene/protein identifier is extracted from the article, the sequence is obtained and BLAST is performed using the sequence as a query (16) against the tomato genome sequence (the latest build). Once the gene model corresponding to the one reported in the literature has been identified correctly, the curator enters the reported synonyms for the identified locus in the corresponding locus detail page.
Gene–gene associations
Gene to gene associations are made using custom tools on the locus detail page. Two genes can be associated through a relationship term describing the nature of the association. A list of gene relationship terms was created in-house at SGN based on the needs of the curators. These relationships can be grouped into orthologuous relationships, describing related genes across different organisms, or paralogous relationships, describing related genes within an organism. Other relationships that were defined include physical interactions (protein complex), co-expression, interaction, activation, inhibition, regulation and pairs of directional relationships to describe the positions in regulatory and other networks (upstream and downstream). When curating from a locus page, an associated locus and the type of relationship must be selected, and optionally a supporting reference can be added from the list of publications linked with the locus. Analogous to Gene Ontology (GO) annotations, evidence codes are assigned as appropriate for the publication at hand, for example, ‘inferred from sequence or structural similarity’.
Ontology development and use
The highest-quality functional annotations are created manually through the curation of the literature and the use of ontologies. SGN curators are active participants in the development of the Plant Ontology (PO) (17) and the SGN database relies heavily on ontologies such as the Phenotype and Trait Ontology (PATO) (18), and GO (19). In addition, the Solanaceae-specific ontology called the Solanaceae Phenotype Ontology (SPO) has been developed at SGN. The curation interface allows curators the ability to associate any GO, PO and SPO term to any locus, allele or accessions. Additional fields can include annotation details about alleles, accessions, lines, populations, images and biochemical reactions catalyzed by the gene product. Evidence codes and literature citations are curated for most of these fields. The edits are made using the community curation system developed at SGN (3). To cover information from gene to phenotype, the curators make combined use of all these ontologies during curation. Integrating these ontologies into a single interface in SGN provides diverse users the ability to search for terms that carry annotations of genomic and genetic elements. These annotations are periodically submitted to the PO and GO databases.
Results
Multigene family curation
Gene families are groups of genes that encode proteins with similar sequences through the entire length or at specific domains owing to common ancestry. Two flavors of multigene families exist in the SGN database: computationally predicted gene families and manually curated gene families. The procedures chosen to perform automated sequence curation determine how well the gene families are represented in the sequence database. The locus detail page also has information about computationally derived information such as domains and genome location, as well as data on literature references, ontology-based annotations and links to accessions harboring mutations in that locus.
Gene families can consist of two members to several dozen genes arising from a common ancestor. Some examples of curated gene families in SGN include WRKY transcription factors (84 members) (20), Dicer-like (23 members) (21), RNA-directed RNA polymerase (12 members) (22) and auxin-regulated indole acetic acid (IAA) (45 members) (23). Broadly, curation of gene families proceeds along three connected and overlapping approaches: (i) capture of gene family distribution, (ii) populating the functional information and (iii) relationship and network connections of gene families.
Genome distribution of gene families
The order of genes in a chromosome sequence is the result of a number of complex gene rearrangement events involving gene loss and gene duplications including partial and whole genome duplications (24). Visualization of gene positions in the genome is therefore important for the interpretation of genome structure and evolution. Comparative analysis of the distribution of two gene families, SlWRKY and SlSAUR, from tomato (Heinz cultivar) with the sequences of the Solanum galapagense and S. pimpinellifolium genomes, revealed that they are almost identical in cultivated and wild accessions (Strickler et al., unpublished results). Individual members of gene families can be clustered or distributed all over the entire genome (7, 8, 25). An example is shown in Figure 1.
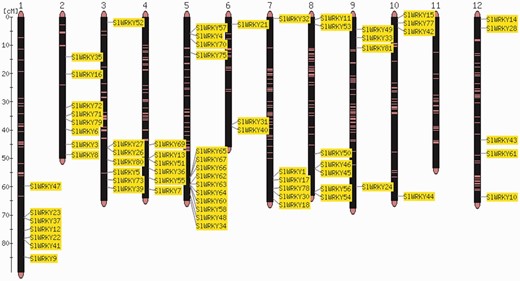
Map viewer. Chromosome distribution of the 82 members of the tomato WRKY gene family. The shown locations are based on the physical position of each corresponding genome gene model, based on the ITAG2.3 annotation of the tomato genome.
Annotated gene families as positional markers on chromosomes
The annotation of chromosomal locations of genomic features based on experimental evidence from the literature is one of the primary curational tasks. The recently published genome sequences of tomato (2) and potato (9) have shed new light on chromosomal organization in the Solanaceae family, and provide insights into the evolution of plant genomes (26). In earlier studies on sequence analysis of Solanaceae gene families, P450 mono-oxygenases and serine threonine protein kinases (27) were found to be overrepresented in potato as compared with tomato, and in both plants, the P450 genes were expanded much more than in Arabidopsis thaliana. Confirmation of computationally identified gene families with experimental data adds the next layer of annotation, and these genes can serve as significant anchors in the genome sequence for researchers looking for unknown genes located near the experimentally validated genes. Along with micro-synteny, the conserved order of genes and gene families across organisms (28) are important data types for genome comparisons. On SGN, gene families can be directly visualized on the comparative mapviewer (29).
Annotation of functional information to gene families
Functional annotation of genes based on gene family information can increase curation efficiency significantly because many of the gene family members will share some of the functional characteristics, and if the appropriate user interfaces are available, pertinent annotations can be propagated to all members of the family in one editing step. In the SGN system, the gene family detail page allows ontology-based annotations on a gene family basis, which are then propagated to each member of the family. Each member of the gene family will also have specific characteristics that do not apply to other members of the gene families. Such annotations can be entered on the locus detail pages with the traditional locus curation interface.
A consistent nomenclature is important for searching and identifying gene families. The gene family name as cited in the literature is curated, while the names of the computational annotation from the sequence curation pipeline of gene families is also retained as synonyms (usually representing some characteristic domain of the gene family example, Basic helix loop helix (bHLH), DNA-binding, WRKY domains, etc). More detailed descriptions of gene activity, synonyms and specific ontology annotations are directly made on the locus detail page.
Loci can be manually associated with a gene family using the ‘Associated Loci’ section on the Locus detail page. The SGN software allows users to visualize the relationships of annotated gene families, such as paralogs and orthologs, facilitating genome comparisons and the study of genome organization (30). Paralogous relationship between genes within the same species and orthologous relationships across species can be visualized as networks in the ‘Graphical View’ section, also found on the locus detail page.
Gene networks
In addition to membership in gene families, the ‘Associated Loci’ also enables the association of more extensive functional relationships between genes, such as information of interactions in pathways (e.g. a gene upstream or downstream of another gene, involvement in regulation, inhibition or co-expression). Each relationship is curated along with the appropriate evidence codes and literature citation from where the information was extracted.
A number of research articles reporting genome-wide profiling of complex data, such as gene families, using a wide range of new technologies, were published following the release of the tomato genome sequence. Examples of such articles published in the year 2012 include attempts to improve resolution of association mapping using genome-wide studies of quantitative trait loci of an admixture of cultivated cherry tomato and the wild ancestor S. pimpinellifolium (31), and genome-wide ortholog searches for tissue-specific genes and promoters across species (32). Several articles report on genome organization and chromosomal rearrangements in a cross-species comparative context (33). Others report on genetic and genome-wide transcriptomic analyses identifying genes (34), genome-wide analysis of gene regulatory elements like WRKY, Dicer-like Argonaute, RNA-dependent RNA polymerases (21) and Aux/IAA (23), in addition to a significant number of articles on genome-wide microarray analysis and gene expression studies in Solanaceae species (35).
A goal for genome databases is the ability to display complex biological information in an accurate, up-to-date and comprehensive manner. In most model organism databases, the gene detail pages, usually covering just one single gene, are the primary means of conveying annotated genomic information. The single gene page approach is limiting, as users want access to all orthologous and paralogous genes, and the biological networks that the gene families are associated with, allowing for far more efficient comparisons within and between species.
Several genome databases comparing gene families between species already exist (5, 6). However, most of these sites focus specifically on automated builds of gene families, without the ability to curate the gene family information, and do not integrate the gene information with other data types such as phenotypes. In the SGN database, data types such as genetic, genomic and phenotypic information are tightly integrated with gene families, allowing users to explore the biology of a given family in a comprehensive manner.
Curation interface
The curation interface created for gene family curation is based on the regular SGN community curation interface (3), with improvements to facilitate gene family search and gene family curation tools (Figures 2 and 3). The system is linked to the SGN alignment analyzer and the SGN mapviewer (29).
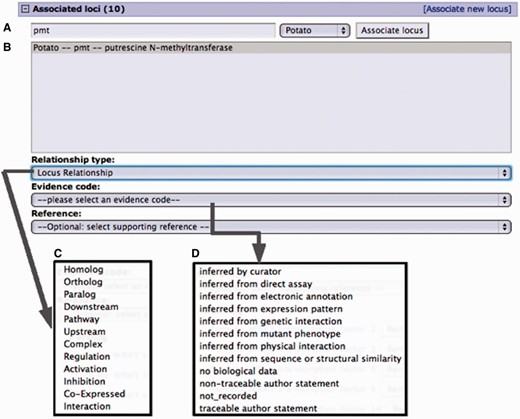
Curation interface for associating a locus from an existing locus page. (A) A search for a locus name by the organism common name. (B) Select locus from the result list pop-up menu. (C) Select the relationship type from the pop-up menu. (D) Select evidence code for the described locus relationship. Adding a reference is optional. Only references associated with the loci involved are presented in the pop-up menu.

Curated gene family page. A family of putrescine N-methyltransferase (PMT) homologous genes from multiple Solanaceae organisms. Gene family details are editable, and curators can add members to the family in a similar manner as associating loci from the locus page, except for the relationship type, which is predefined. Gene family members are listed by organism with an evidence code.
Visualizing networks of genes and multigene families annotated to drought stress
Here, we use drought response as an example of how gene family and gene networks are annotated. Plant response to biotic and abiotic stress is the result of complex synchronized actions of gene networks (36). Drought is a world-wide problem, and understanding the processes underlying drought stress tolerance in plants is a high priority in many research projects. Drought resistance is a complex process, and little is known about the molecular mechanisms underlying the plant response and tolerance (37). The responses involve biochemical, physiological, molecular, cellular and whole-plant changes (38). Genetic, molecular and genomic analyses of drought response and tolerance in a number of plants such as Arabidopsis (39), rice (40), maize (41), tomato (20) and other plants have revealed several drought-inducible genes that appear to play different roles in managing drought stress. Breeders and researchers look for specific types of information in biological databases regarding candidate genes that may be exploited for crop improvement (37). Recent understanding of regulatory networks of drought response allows for developing practical strategies for engineering drought-tolerant varieties (38). High-throughput studies in tomato with drought-tolerant Solanum pennellii introgression lines identified nearly 400 genes found to be responsive to drought (42). The Solanaceae provide a great model, as S. lycopersicum is a drought-sensitive species, while the wild relative S. pennelli is a drought-resistant species.
Using curated information annotated to the GO term ‘response to water deprivation’, we attempted to visualize the genes that have been assayed for drought tolerance. This was combined with other annotated information including their roles in regulatory, signaling and biochemical pathways, and involvement in developmental and growth processes studied under drought stress. These genes have different functional roles: some are transcription factors (WRKY, bHLH), and others are signaling proteins. The network diagram was created by using Cytoscape (43). Nearly 260 genes were found to play a role in drought stress/tolerance. Figure 4 gives a network visualization of these genes. A few of the gene families that figure prominently in these studies include SlWRKY and SlSAUR genes. Additionally, some genes were linked to accessions and introgression lines of tomato such as Introgression (IL)1-1 and IL2-1, which are known to be drought-resistant lines. It is now possible to visualize networks of genes, pathways and phenotypes, which allow users to perform in-depth studies aimed at unraveling the molecular machinery underlying the phenotypes known to be drought resistant.
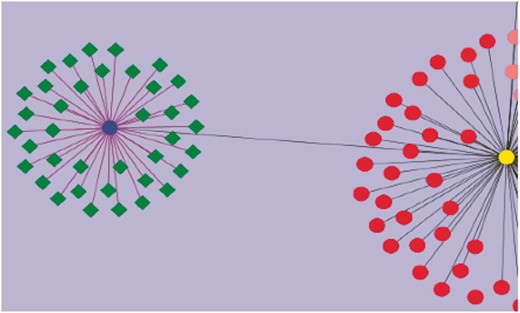
Visualization of genes associated with drought tolerance in Solanaceae. A simple example network is shown. The nodes represent genes, and the edges represent relationships between the genes. The blue circular node is WRKY 39 and the green nodes having diamond shapes are genes associated with drought, annotated in the SGN database. The purple edges connecting WRKY 39 to the other genes represent the relationship based on the evidence code ‘Co-expressed’. These genes have been curated from an article reporting a transcriptomic study of drought response genes in tomato species. The deep gray colored edge connects WRKY 39 transcription factor to WRKY 1 (yellow circular node) in a paralogous relationship. Similarly all red colored nodes are the WRKY gene family members curated in SGN. This network diagram shows the genes in the SGN database that have been currently annotated to the GO term, ‘GO: response to water deprivation’.
Discussion
The era of cost-effective next-generation sequencing technologies has enabled the rapid sequencing of a large number of plant genomes, but as a consequence, also has caused a move away from model species. These large consortia had significant curation and annotation efforts, as typified by the genome projects of model organisms such as Arabidopsis and Oryza, which are not available to newly sequenced genomes. As limited resources are now being spread thinner, comparative genomics depends on increasing the efficiency and quality of manual curation. The curation at SGN, a clade-oriented database for Solanaceae species, has been based on a community-based paradigm for many years. The community curation approach has been well accepted by the Solanaceae researchers, and the number of new community curators from the research community continues to expand (3). Currently, there are 640 loci under the control of 135 locus editors. Of the almost 35 000 tomato loci in the database, <2000 have any associated data such as literature or Genbank accessions (15). Since the release of the tomato genome sequence in 2012 we expect these numbers to rise quickly, with the entire genome sequence being available, researchers can easily investigate complete gene families. Examples of such genome-wide profiles of gene families that we curated are the SlPIN, SlWRKY, SlSAUR families; such articles report genes that can sometimes number in the hundreds. Developing curation tools that can capture and then automatically populate many fields such as GO and PO annotations, cellular and tissue location, growth and developmental stages to all of members of a gene family represents an important strategy that assures high data accuracy while saving curation time and resources. Similarly, we could populate the phenotype information of genes that have been studied using specific accessions and lines; for example, SlWRKY genes were being assayed from the drought-tolerant introgression lines IL1-1 and IL2-5, and several other genes assayed for drought tolerance from one study has used the accessions LA4040 and LA0197.
Gene family and gene network curation depend on high-quality structural annotations, which often have to be manually curated. Processes for curation of structural and functional annotations on the gene level are a prerequisite for curation of gene families and gene networks.
To the research community at large, high-quality gene family annotations are important tools for candidate gene approaches, biotechnological engineering and comparative and evolutionary studies. Re-sequencing of accessions and lines depends heavily on the annotation quality of the reference genome used in the annotation pipeline. In such cases, the specific and well-annotated gene families of any one of the lines can be used to map and analyze the new genomes.
The ability to curate gene networks for regulatory and other types of pathways will allow researchers to interrogate the database with more powerful queries.
Future directions
In future versions, the integration of manual and automated gene family builds will be improved. A curator should be able to import automated gene family builds into the manually curated gene family space for further refinement. The visualization of families and networks will be implemented based on standard programs such as Cytoscape (43) and more robust queries for gene networks will be developed.
Funding
This work was partially funded by the National Science Foundation (DBI-0820612).
Conflict of interest. None declared.
References
Author notes
Citation details: Pujar,A., Menda,N., Bombarely,A., et al. From manual curation to visualization of gene families and networks across Solanaceae plant species. Database (2013) Vol. 2013: article ID bat028; doi:10.1093/database/bat028

{kind=link}
{kind=link}
{kind=link}
{kind=link}