





Abstract
Comprehensive disease classification, integration and annotation are crucial for biomedical discovery. At present, disease compilation is incomplete, heterogeneous and often lacking systematic inquiry mechanisms. We introduce MalaCards, an integrated database of human maladies and their annotations, modeled on the architecture and strategy of the GeneCards database of human genes. MalaCards mines and merges 44 data sources to generate a computerized card for each of 16 919 human diseases. Each MalaCard contains disease-specific prioritized annotations, as well as inter-disease connections, empowered by the GeneCards relational database, its searches and GeneDecks set analyses. First, we generate a disease list from 15 ranked sources, using disease-name unification heuristics. Next, we use four schemes to populate MalaCards sections: (i) directly interrogating disease resources, to establish integrated disease names, synonyms, summaries, drugs/therapeutics, clinical features, genetic tests and anatomical context; (ii) searching GeneCards for related publications, and for associated genes with corresponding relevance scores; (iii) analyzing disease-associated gene sets in GeneDecks to yield affiliated pathways, phenotypes, compounds and GO terms, sorted by a composite relevance score and presented with GeneCards links; and (iv) searching within MalaCards itself, e.g. for additional related diseases and anatomical context. The latter forms the basis for the construction of a disease network, based on shared MalaCards annotations, embodying associations based on etiology, clinical features and clinical conditions. This broadly disposed network has a power-law degree distribution, suggesting that this might be an inherent property of such networks. Work in progress includes hierarchical malady classification, ontological mapping and disease set analyses, striving to make MalaCards an even more effective tool for biomedical research.
Database URL:http://www.malacards.org/
Introduction
One of the greatest challenges of biomedical research is deciphering the underlying mechanisms of human diseases, which requires accurate classification and annotation. Most human diseases arise due to complex interactions between multiple genetic variants and environmental risk factors (1); thus, studying diseases could shed light on basic biological mechanisms. Diagnosis and treatment are facilitated by the huge amount of information coming from genomics and proteomics research, allowing molecular level support for medical decisions. The integration of these massive amounts of information under a single disease nomenclature is an enormous challenge.
Our survey has identified >60 disease-related databases. Each of these focuses on different aspects of disease annotation and/or contains a partial specialized list. For instance, Online Mendelian Inheritance in Man (OMIM) initially focused on monogenic disorders; in recent years, it has been expanded to include complex traits and the associated genetic mutations that confer their susceptibility (2, 3). PharmGKB specializes in how genetic variation is related to drug response (4). The toxicogenomics database CTD stores information about the effect of environmental chemicals on human health (5). Disease Ontology aims to supply a cross-referenced formal semantically computable structure of all diseases (6). GeneTests provides authoritative information on genetic testing (7).
The different databases use diverse terminologies. For example, ‘usher syndrome’ is also called ‘retinitis pigmentosa-deafness syndrome’, ‘Graefe-Usher syndrome’, ‘dystrophia retinae pigmentosa-dysostosis syndrome’, ‘deafness-retinitis pigmentosa syndrome’ or ‘Hallgren syndrome’ in the various sources; we have not found any particular resource that portrays the fact that all of these are aliases for the same condition.
Promising attempts to settle the varied disease nomenclature are presented via knowledge representation through standardized vocabularies, to ensure both effective information sharing and interoperability among information systems (8,9). There are several vocabularies, ranging from class-specific ones, such as the National Cancer Institute (NCI) Dictionary of Cancer Terms (http://www.cancer.gov/dictionary), NCI Drug Dictionary (http://www.cancer.gov/drugdictionary) and the Infectious Disease Ontology (IDO) (http://infectiousdiseaseontology.org/page/Main_Page), to more broadly disposed ones, such as the International Classification of Diseases (ICD) (10, 11), the Unified Medical Language System (UMLS) (12), the Systematized Nomenclature of Medicine—Clinical Terms (SNOMED-CT) (13), the Medical Subject Headings (MeSH) (http://www.nlm.nih.gov/mesh/) and the Disease Ontology (DO) (6). Such data structures range from flat lists, such as OMIM, to hierarchies, as exemplified by the DO. However, significant inconsistencies prevail in basic terms pertaining to diseases, diagnoses and clinical phenotypes. Some vocabularies attempt to map existing terminologies to each other. Terms in DO are often linked to well-adopted terminologies, such as SNOMED-CT, ICD-9 and ICD-10, MeSH and UMLS, but the disease concepts contain limited annotations on each disease (8). The merged disease vocabulary (MEDIC) attempts to integrate OMIM terms, synonyms and identifiers with MeSH terms, synonyms, definitions, identifiers and hierarchical relationships (14). Nevertheless, existing vocabularies are only partially cross-connected to each other, and they do not define disease concepts uniformly. Moreover, most existing disease databases do not associate their diseases with any ontology, or only to some of them, which greatly limits the effectiveness of such formalizations in supplying unifying disease definitions.
Notably, existing resources are also characterized by heterogeneous navigation, architecture and querying mechanisms, often with complicated usability, requiring non–trivial-specific knowledge to obtain the actual information. Hence, integration of different databases is sorely needed to allow a comprehensive view of biomedical disease knowledge, to support clinical and basic research. This integration must encompass all types of diseases, include coherent nomenclature, unified annotation and a friendly user-interface easily accessible to both medical and scientific professionals.
To address this challenge, we have compiled MalaCards, a comprehensive human disease compendium, currently unifying 44 disease sources into a convenient format of ‘disease cards’, each integrating relevant information and listing numerous known aliases for each disease, along with a variety of annotations as described later in the text. MalaCards inputs range from text-mined to manually curated data sets. The database is compiled by an automatic computational information retrieval engine, which populates annotated disease cards, using remote data, as well as information gleaned using the GeneCards platform (15). MalaCards covers ∼17 000 human diseases. Its integrative generation and annotations, links to GeneCards, comprehensive search and user-friendly interface make it an effective tool for researchers and clinicians.
MalaCards disease list
Integrated disease list generation
An offline process is responsible for generating a comprehensive-integrated list of diseases by mining heterogeneous, partially overlapping sources (Supplementary Table S1), unifying names and acronyms. We have implemented an automatic disease name unification algorithm, which strives to transform each mined disease name to a canonical form, while simultaneously retaining the original form for the alias list. This canonical form is constructed by a series of steps that strip the non-informative components to enable textual comparison, as follows:
Names are converted to lowercase, and non-alphanumeric characters are removed. Next, descriptive words like ‘disease’, ‘syndrome’, ‘deficiency’, ‘failure’, ‘type’, as well as conjunctions, articles and prepositions, are stripped. The key for deciding whether a word should be removed is whether two names for the same disease differ by that word alone, for example, ‘Werner syndrome 1’ versus ‘Werner 1’ or ‘Alzheimer’ versus ‘Alzheimer disease’.
Equivalent words are merged, like ‘juvenile’ and ‘childhood’, ‘kidney’ and ‘renal’, ‘fast’ and ‘rapid’, as well as different numbering formats, such as Roman versus Indian/Arabic, so that ‘Glycogen Storage Type IV’ coming from DO, OMIM and diseasecard is united with ‘Glycogen Storage Disease Type 4’ coming from NIH Rare Diseases and GeneTests.
Plural/singular and possessives are handled, e.g. ‘s’, ‘ies’, ‘y’, ‘es’, ‘’s’ to unite ‘Refsum disease’ coming from numerous sources with ‘Refsum’s disease’ coming from DO, and ‘Neurodegenerative disease’ coming from DO with ‘Neurodegenerative diseases’ coming from Novoseek.
Word stemming, using the porter stemming algorithm (16), is applied to each word. Stemming is used to reduce inflected or derived words to their stem, base or root form. It is usually sufficient that related words map to the same stem, even if this stem is not in itself a valid root. For example ‘Alzheimer’ and ‘Alzheimer’s’ will be mapped to the same root ‘alzheim’.
In an intermediate step, spaces are removed and canonical forms are compared, as a first decision point for unification. This unifies cases such as ‘Leukoencephalopathy with Brain Stem and Spinal Cord Involvement …’ coming from GeneReviews and OMIM with ‘Leukoencephalopathy with Brainstem and Spinal Cord Involvement …’ coming from GeneTests and Genetics Home Reference.
Stemmed words are lexically sorted to account for different order of words in the same name, such as in ‘Common Variable Immunodeficiency’ coming from DO, NIH Rare Diseases, Genetics Home Reference, DISEASES and NovoSeek compared with ‘Immunodeficiency, Common Variable’ coming from Genetics Home Reference.
The canonical form is then hashed and used for comparison with transformed new names.
Importantly, the lexical manipulations are done solely for the purpose of canonicalization. On decision on merging, all aliases of all forms are kept with the corresponding attribution of their sources, so that they are visible on MalaCards, as well as searchable.
Sources are evaluated based on a pre-defined order of priorities (Supplementary Table S1). Each name is transformed and compared with the existing list of names. If the name already exists, this entry and its associated aliases and source annotations are added. If the name does not exist, it is added to the list as a new disease. The name list serves as the basis for the generation of diseases within the MalaCards database. Importantly, following this process, a manual curation process removes erroneous diseases from the list.
The name integration process mines 85 377 disease names and aliases, belonging to 45 427 distinct diseases as defined by the partial integration within 15 mined name sources. The name unification step performs further consolidation to yield a unified list of 16 919 disease entries, each constituting a MalaCards ‘card’. Notably, these originate from 15 sources, the largest of which supplies ∼3000–6000 distinct entries, suggesting effective disease amalgamation, but at the same time indicating a measure of naming promiscuity (see ‘Discussion’ section).
Disease type grouping
A specific type of hierarchy was introduced, addressing cases of disease names that are identical except for type specification. This is exemplified by Alzheimer’s Disease, for which another 16 MalaCards have the same base name, such as ‘alzheimer disease type 3’ or ‘Alzheimer disease 13’. In total, ∼3000 diseases are thus reducible to ∼700 families. The disease in each ‘family’ having the highest MalaCards information score (MIFTS, see ‘Scoring’ section) is designated ‘parent’. In the case of a tie, priority is given to the disease associated with the highest number of sources, and then to the one with the shortest name. All other family-affiliated diseases are designated ‘child’. The parent/child attributes are labeled ‘P’ and ‘c’ in search results, and all relationships are listed at the top of the ‘Related Diseases’ section.
Disease annotation processes
Annotation schemes
MalaCards has numerous independent disease sources, but it also generates disease-specific information based on gene–disease relations within GeneCards. MalaCards uses four different annotation schemes, as follows:
Source mining
Mining data sources for disease-specific information is used to populate relevant sections of a MalaCard. To this end, we define two types of sources (Supplementary Table S1). Primary sources are those that are used to derive main disease names; some of them also supply annotations (15 sources). Secondary sources are those from which only annotations, aliases, and/or external IDs to existing diseases are derived (29 sources). These sources generally contain non-disease terms intermixed with disease information. Direct source mining provides information for the aliases and descriptions, summaries, clinical features, drugs and therapeutics, genetic tests and anatomical context sections. When appropriate, in-house analysis is performed, to link annotations to diseases, or to integrate and display disease-specific data. For example, we have developed a process that uses UMLS concepts to map diseases to drugs used for its treatment (see ‘The Structure of a MalaCard’ section).
GeneCards search
One central annotation source for MalaCards is the automated use of the GeneCards search engine, including section-specific advanced searches. For example, all of the genes associated with a disease are obtained by using the disease name as a search string, which allows the generation of the related genes section in MalaCards. Importantly, gene association does not imply causality between the gene and the disease. Associations sometimes include annotation like ‘unaffected’, and this can be verified using the ‘GeneCards section context’ link. Similarly, the publications associated with a disease are obtained via a search for its name in all of the publication titles within GeneCards.
GeneDecks set analyses
MalaCards implements a strategy in which gene–disease relationships within GeneCards are used to create disease-specific content. For this, we leverage GeneCards’ GeneDecks tool (17), in its Set Distiller mode. The disease-associated gene set (generated as described earlier in the text) is forwarded to GeneDecks, which distills statistically significant descriptors enriched in this set. For example, in the ‘Atherosclerosis’ MalaCard, ‘cardiovascular system’ is thus entered into the phenotypes section, whereas ‘apoptosis’ into the pathways section. This process also assigns a relevance score for every hit and is used to populate the related diseases, phenotypes, pathways, compounds and GO terms sections. In these sections, the relevant tables display the affiliating genes, linked to their respective contexts within GeneCards.
MalaCards search
We use MalaCards searches to populate additional sections, including elucidating new relations among diseases in the related diseases section and associating tissues in the anatomical context section.
Scoring
MalaCards assigns five types of scores:
- MalaCards composite relevance score (MCRS). Assigned to descriptors provided by the GeneDecks set analyses mechanism (Figure 1). The score is defined as:where: SGD is the rank of the GeneDecks score, which orders descriptors first by their GeneDecks P-value and then by the size of the group of genes associated with the descriptor. SLR (i) is the Solr search engine score’s rank of a gene shared between the descriptor and the disease. Ns is the number of data sources supporting the descriptor. Thus, the score takes into account the hit importance in GeneCards, the significance of the specific attribute according to GeneDecks, as well as the number of supporting sources.
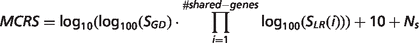
GeneCards search relevance score (GSRS). Obtained by the Solr-based GeneCards search engine (http://www.genecards.org/index.php?path=/HTML/page/searchHelp#relevance). This relevance score takes into account the number of hits, and the importance of the fields in which they were found.
MalaCards search relevance score (MSRS). Obtained by the Solr-based MalaCards search engine, as described later in the text.
MalaCards information score (MIFTS). Assigned to each disease by summing the base 10 logarithms of the counts of its populated annotations. MIFTS defines the richness of information in each card. This score currently ranges from 1 to 101.
MalaCards composite-related diseases score (MCRDS). Assigned to entries in the related diseases section. It is computed as the sum of the MCRS and the MSRS. Before this, each of these two score values is normalized by equating the means as well as the standard deviations for the two distributions across all of MalaCards. A bonus amounting to the average of the two scores is added to diseases coming from both GeneDecks set analysis and MalaCards search.
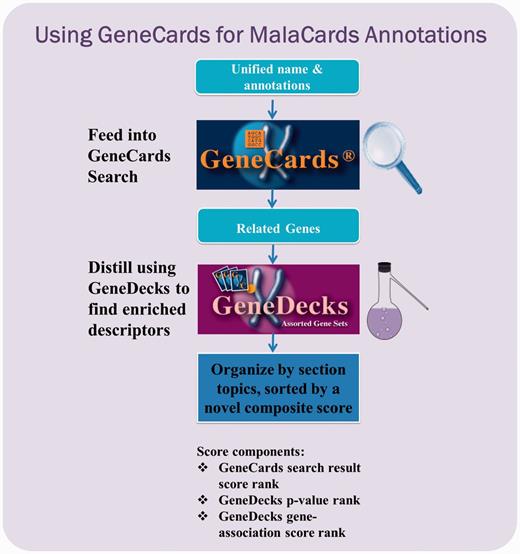
GeneCards-based annotation pipeline. Each unified disease name is fed into the GeneCards search engine to find its associated gene set, as well as publications, disease–gene associations and the corresponding contexts wherein the match occurred. The set is then forwarded to GeneDecks, which distills statistically significant descriptors (e.g. ‘cardiovascular system phenotype’, ‘apoptosis’) for the genes in the set. These shared descriptors, sorted by relevance, are featured in various MalaCards sections.
The structure of a MalaCard
Each MalaCard is composed of 15 sections (Figure 2). For each section, the left-hand side panel shows the contributing sources, with links to their home pages. Superscripts in the main panel (right side) denote and deep-link to disease-specific information within the sources, where available. The section-source mapping is specified in Supplementary Table S1.
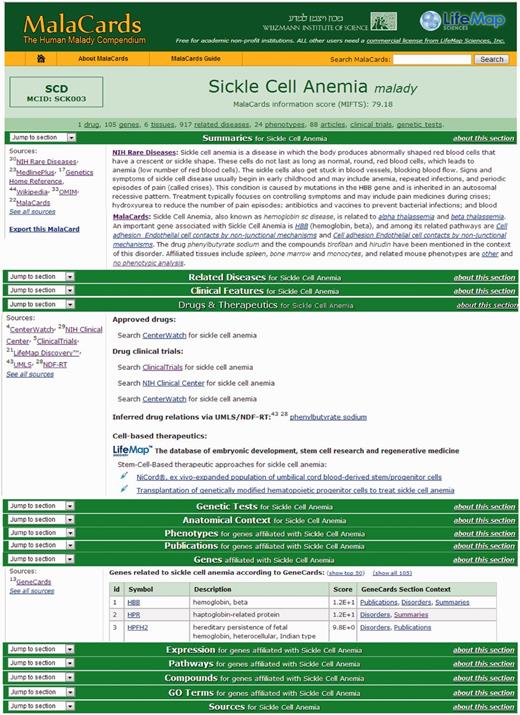
MalaCards sections. Subset of the MalaCard for sickle cell anemia. The left-hand side of each section lists its contributing sources. The right-hand side contains nuggets of section-related information, with deep links to the original sources for comprehensive scrutiny. 9. A ‘stats bar' containing the statistics of a selected set of populated sections is displayed in the card header.
Header
Displays the disease name and acronym (where available). The name is assigned according to the highest source in the names hierarchy (Supplementary Table S1). Acronyms are either supplied by specific sources, such as NCBI Bookshelf and Wikipedia (currently totaling 120 cases), or taken to be the shortest disease name/alias, provided that it is five characters or less (another 1260 cases). We note that acronyms may not be unique, e.g. both Williams Syndrome and Werner Syndrome have the acronym WS. Also shown is an in-house–generated unique and stable MalaCards ID, constituting the first letter and subsequent two consonants of the disease name, followed by a three digit serial number. For example, the symbol for ‘Sickle Cell Anemia’ is SCK003. The header also features the ‘stats bar’, containing the statistics of populated annotations for select sections, where available.
Aliases and descriptions
These are extracted from a subset of the sources (Supplementary Table S1), according to the unification algorithm described earlier in the text. Strongly similar aliases, even if trivially different, are included, to match common expectations and to facilitate searches. The disease name appears first, with its own associated source-indicating superscripts. The alias list is sorted first by the count of contributing sources and then sub-sorted by descending length. The number of aliases for a single disease currently reaches as high as 33. This section also includes the sub-section ‘External IDs’, which lists identifiers from external databases relevant to this disease, such as MeSH, ICD9 and SNOMED-CT. These IDs are searchable, and they allow cross-referencing between MalaCards and other databases.
Summaries
This section displays information about the disease, as extracted from a subset of the sources (Supplementary Table S1). A summary typically includes a short definition of the disease, organs involved, etiology and main symptoms. One of the summaries is an automated MalaCards-generated summary, highlighting the main annotations in the specific card.
Related diseases
The top of the section displays the disease type classification if available. Related diseases are obtained in two ways: first, by GeneDecks set analysis, whereby other diseases computed to have significant shared descriptors for the target disease’s-related genes are displayed. Second, as matched by MalaCards searches. The related diseases obtained are prioritized by a MalaCards composite-related diseases score. This section also includes a network image displaying the top 20 related diseases and their interconnection. Currently, edge distances between the MalaCard’s disease and the other nodes are not significant.
Clinical features
It provides information and links about symptoms and other clinical attributes, according to OMIM and DO (Supplementary Table S1). Symptoms typically represent changes from normal function, sensation or appearance, but they may also be disease names independently defined in MalaCards.
Drugs and therapeutics
It contains information regarding both drugs and clinical trials. Drug information is obtained in two ways. The first method combines information from the UMLS and the National Drug File—Reference Terminology (NDF-RT). Initially, a MalaCards name is mapped to a UMLS concept representing a disease by using the MetaMap system (18,19). Subsequently, the NDF-RT within UMLS is used to provide a link of such disease concepts to drug(s) via the ‘may be treated by’ relationship. In the second method, drug information is supplied through a disease-specific search link to CenterWatch for newly approved drugs. CenterWatch is also a source for clinical trials data, also obtained via deep links for searching ClinicalTrials.gov and the NIH Clinical Center (Supplementary Table S1). Additionally, this section presents cell-based therapeutics approaches from LifeMap Sciences, linking specific cell lines to the disease as candidate therapeutic approaches.
Genetic tests
It provides descriptions of genetic testing, specialized cytogenetic testing and biochemical testing for inherited disorders. These are extracted from GeneTests (Supplementary Table S1). The section shows both clinical and research laboratories performing genetic tests (7).
Anatomical context
It displays disease-related cell types, anatomical compartments and organs, as well as related in vitro cell types from human and mouse. These data are derived from LifeMap Discovery™, the database of embryonic development, stem cell research and regenerative medicine (http://discovery.lifemapsc.com/). Also displayed are MalaCards organs/tissues related to the disease. These are obtained by the MalaCards search mechanism applied on a pre-defined list of organs/tissues.
Phenotypes
It provides murine phenotypes, which are obtained from MGI (Supplementary Table S1) as contextually related to the target disease using the GeneDecks set analysis. Phenotypes are scored according to their relevance using the MCRS, and they are deep-linked to their sources.
Publications
It provides scientific articles associated with the disease, obtained from PubMed (Supplementary Table S1) by the GeneCards search mechanism. Matched articles are ranked sequentially according to the number of sources that associate the article with the disease-related genes in GeneCards, by date of publication, and according to the individual source scores for article/gene relationships.
Affiliated genes
It provides the list of affiliated genes found by searching GeneCards. The table shows gene symbols, descriptions and a deep linked GeneCards section in which the disease association occurs. A relevance score is also shown, as computed by the GeneCards search engine. More than half of the MalaCards entries (9353, 53%) are associated with genes, with only ∼4900 of them associated with OMIM genetic disorders. Importantly, cases of circular gene–disease association have been removed. These are identified as instances in which a gene is linked to a MalaCard based on a disease connection in GeneCards that arises from GeneCards’ use of MalaCards as an only relevant source.
Expression
It provides normal tissue mRNA expression intensity charts from BioGPS for genes related to the disease. Each tissue-related column shows color-coded values for up to 100 of the most highly expressed genes, ranked by their expression level. Only the top 20% chart-wide expression values are shown, thus highlighting the tissues in which the disease-related genes are most highly expressed.
Pathways
It provides pathways related to the disease, obtained by GeneDecks set analysis. The pathways are extracted from a subset of the sources (Supplementary Table S1). Entries are scored according to their relevance using the MCRS. As pathways are extracted from several sources, partially or fully overlapping pathways may be displayed. A GeneCards pathways integration effort is underway; once deployed, its results will be displayed in MalaCards as well.
Compounds
It provides relationships between MalaCards diseases and chemical compounds (small molecules, metabolites) obtained by GeneDecks set analysis. Information sources are as shown in Supplementary Table S1. Entries are scored according to their relevance using the MCRS.
GO terms
It provides gene ontologies (cellular component, biological process and molecular function) obtained by GeneDecks set analysis from Gene Ontology (Supplementary Table S1). The table displays the ontology name, GO identifier (deep-linked to the GO entry) and the implicating genes, linked to GeneCards. The entries are scored according to their relevance, using the MCRS.
Sources
This section provides links to the collection of MalaCards sources (Supplementary Table S1).
MalaCards search and browsing
The main search facility of MalaCards is within its home page (Figure 3A), which also provides links to a sample disease and its various sections. The home page further allows one to view a random malady and harbors a useful browsing tool accompanying an alphabetic disease index. Also portrayed are links to GeneCards and its suite members (e.g. GeneDecks, GeneALaCart and GeneLoc) and general MalaCards information and news.

MalaCards home page and search results table. (A) MalaCards 1.03 home page, including search, sample disease, logos and links to GeneCards and associate suite members and a random disease generator. (B) Example of table of search results for the ‘pemphigus’ query. Columns include disease name, MIFTS and relevance score.
MalaCards’ searches use Solr (http://lucene.apache.org/solr/), a publicly available server based on Apache’s Lucene indexing and text search API. Lucene returns a set of scored MalaCards whose annotations contain the search string. The search engine uses standard features, such as Porter stemming (16) and Boolean operators (with AND being the default of multiple search terms). Our tailored features include score boosting of disease names and aliases. Examples can be found in the MalaCards search guide (http://www.malacards.org/pages/searchguide).
Search results include disease name, disease type parent/child association, MIFTS score and relevance score (Figure 3B). The relevance score is as described in the Lucene Similarity class web manual (http://lucene.apache.org). The score encompasses term frequency, term conjunction, inverse document frequency and field length normalization. For ease of interpretation, the displayed score = log2 (Lucene score) +10.
In the specific example shown (Figure 3B), a search for ‘Pemphigus’, a group of rare skin conditions with autoimmune etiology, results in an associated set of conditions, such as variants of this disease (‘Vulgaris’, ‘Foliaceus’), other related skin conditions in terms of clinical presentation [‘Hailey–Hailey disease’ (genetic), ‘Ritter’s disease ‘(infectious)], as well as conditions that are precipitated by the disease, such as ‘Blindness’, that can be caused by scar formation on the eyelids and eyeball, or ‘Gingivitis’.
A user can download the card data to a parsable Excel sheet using the ‘Export this MalaCard’ button on the left hand side of the summaries section. Data can also be obtained from the authors.
MalaCards disease network
MalaCards provides a comprehensive and rich source of disease annotation and correspondingly, a large number of potential disease–disease associations. This allows the construction of a MalaCards-based network with 16 919 disease nodes, connected through edges occurring if one disease comes up in the MalaCards search of another. Linking nodes by such an annotation metric can capture more information than edges solely representing gene-sharing as previously used (20). We generated such a network via the MalaCards database; a subset is shown in Figure 4A. Nodes and edges are colored corresponding to their cluster association, using the Markov Clustering Algorithm as implemented in the Gephi software (21). Node sizes increase with increasing authority scores, calculated by Gephi’s HITS algorithm (21, 22), which is computed by the sum of the hub values for every outgoing node.
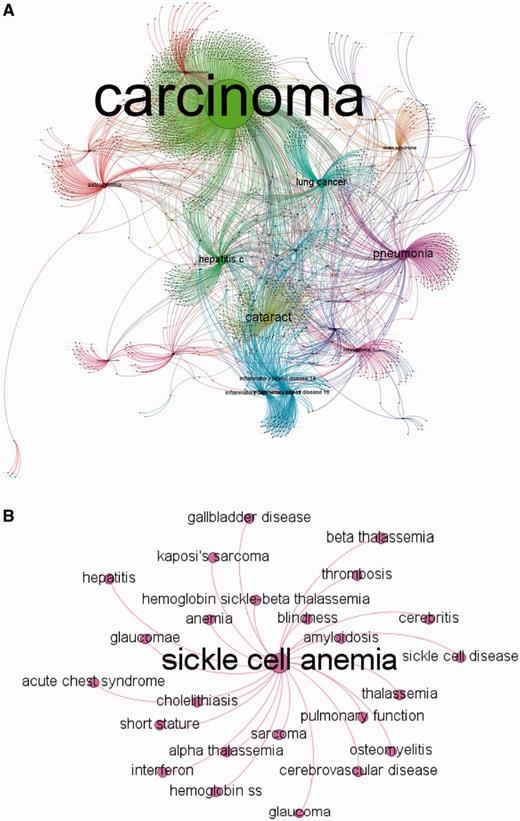
MalaCards disease network. (A) MalaCards disease network created by random sampling of 12% of the nodes, conserving the degree distribution. The network is clustered, whereas nodes and edges are colored according to their cluster association and sized by their authority parameter (22). This figure was produced using Gephi (21). (B) A subset of the directly connected nodes for ‘Sickle Cell Anemia’.
Of ∼1300 diseases that comprise the diseasome network of Goh et al. (20), ∼1200 were mapped to the MalaCards network using the naming unification algorithm described earlier in the text. Goh et al. used a reduced OMIM list of 2929 genetic disorders with strong gene disease association as a departure point, converting it into a ∼50% smaller list by merging disease sub-types of a single disease. The ∼92% one-way overlap between the two disease compendia indicates the validity of MalaCards’ naming procedure. However, the MalaCards network also contains ∼14 500 nodes not present in the OMIM sub-group. Notably, this much larger disease network portrays a power-law in its degree distribution, spanning four orders of magnitude (Figure 5A), as seen for the considerably smaller network of Goh et al. (20).
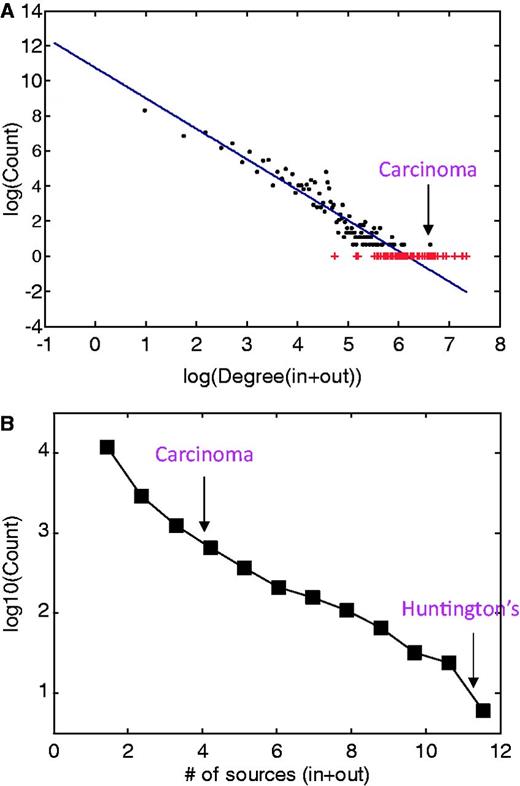
Disease network properties. (A) Disease degree distribution for incoming and outgoing MalaCards search-results edges. The continuous line represents the fit to the log–log binned data, following the function f(x) = ax + b with a = −1.7 and b = 10.8, obtained from a least-square fit with adjusted R2 of 0.9. Outliers are noted in red. (B) Distribution of the number of sources associated with each disease, supplying either or both names and annotations.
A much smaller sub-network (25 nodes) shown in Figure 4B demonstrates that the outgoing links for sickle cell anemia are biologically and clinically meaningful and include other types of anemia and thalassemia. We also observe links to precipitated conditions, such as blindness, which can result from retinal artery occlusion or by classic sickle retinopathy (23), and short stature (24), which is known to occur in children with sickle cell anemia. Interestingly, the degree is not correlated with the number of sources for a given disease (not shown); most diseases come from a single source, whereas some arise from as many as 12 sources (Figure 5B).
Discussion
Automatic data mining
A clear advantage of automatic data mining is rapid extraction of large amounts of data from multiple sources, as well as the use of computerized heuristics. A well-known disadvantage is the extraction of irrelevant data. One example is the extraction of genes that are annotated to be ‘unaffected’ by a specific disease to be associated with this same disease. Another example is extraction of genes related to ‘non-Hodgkin’s lymphoma’ for ‘Hodgkin’s lymphoma’ because the close lexical resemblance between the names of these diseases. Presently, the GeneCards section context allows the user to evaluate the validity of each gene association. In the near future, we will generate a minicards mechanism [as in GeneCards, (15)] that will enable the user to see the hit context associated with each of the search results.
Name integration
The first and crucial step in disease data integration is name integration. MalaCards addresses this challenge by mining 15 name sources containing 85 377 disease names and generating 16 919 disease entries, in an attempt to create an authoritative disease compendium. This takes place through a combination of naming integration performed by the mined sources themselves and in-house automated text processing. The final outcome is our comprehensive human disease digest, with each malady (about half of them gene-associated) represented in a richly annotated web card.
We note that the MalaCards name integration process still requires substantial improvement, to be addressed in future versions, which may account in part for the relatively high number of diseases. Some outstanding challenges are (i) disease sub-types. This is exemplified by currently showing distinct MalaCards for not only the two sub-types ‘Lactate Dehydrogenase-a Deficiency’ and ‘Lactate Dehydrogenase-b Deficiency’, but also for the general disease term ‘Lactate Dehydrogenase Deficiency’. (ii) Disease descriptors. These are cases where both a basic disease name and one with an added descriptor are assigned web cards, exemplified by ‘Acute lymphoblastic leukemia’ and ‘Acute lymphoblastic leukemia, childhood’. Although there are no clear-cut solutions for such issues, we are exploring improvements to the unification algorithm and MalaCards design, among others by introducing hierarchical MalaCards relationships. (iii) Source augmentation. The MalaCards disease list will be enhanced through the inclusion of additional disease sources. Over 30 additional such sources are in our pipeline and more will likely to be found worthy in the future.
Redundancy and multiplicity
MalaCards currently has nearly 17 000 diseases, 3- to 6-fold higher than in other available disease databases. About half of the diseases are associated with genes, and the other half includes non-genetic diseases, such as ‘Lesion of Sciatic Nerve’ and ‘Yusho Disease’. 402 cards are completely empty with a MIFTS score of zero. These are maintained for completeness, and their annotations will likely be populated in future versions.
Admittedly, there is a considerable measure of redundancy in the underlying disease list, but our policy is to include all disease names that were not unified by our naming algorithm as separate MalaCards. Disease inter-relations are then revealed by the ‘related diseases’ definitions. We will make a special effort to improve the naming algorithm in near future versions. One attempt to alleviate redundancy is already implemented, whereby ∼3000 diseases constituting different types of the same malady were grouped into 700 families. These 3000 entries are still kept as separate MalaCards, as in many cases, different types have distinct summaries, related genes and other annotations.
Moreover, different databases might have different policy regarding unification of distinct disease manifestations. For example, diseasecard, Genetic Home reference and Novoseek define ‘Sialidosis’, whereas NIH Rare Diseases and OMIM define both ‘Sialidosis, type II’ and ‘Sialidosis type I’ separately, each having its specific information. Another example is ‘cholestasis’, which in its general form is defined by five different sources, including DO, diseasecard and DISEASES, but it is divided to sub-types by other sources, such as OMIM and NIH Rare Diseases, which define, for example, ‘benign recurrent intrahepatic cholestasis 2’ and ‘benign recurrent intrahepatic cholestasis 1’.
Annotation improvements
In its present form, MalaCards portrays a considerable variety of annotation entries. In our future plans, we strive to improve this compendium’s annotative power, by addressing the following:
Acronyms and symbols. Acronyms are community-provided abbreviations, often not unique but still useful in scientific communication. We plan to improve MalaCards acronyms list both by direct mining from designated acronyms sources and by an algorithm that will automatically generate a presumed acronym and then check its association with the disease, for example, by publication text mining. Disease symbols, akin to gene symbols in being short, unique, mnemonic and stable (25), constitute an independent vast challenge. If implemented, they will certainly contribute to disease annotation, but we note that such an endeavor might necessitate community involvement.
Related genes. Currently, the list of associated genes for each disease is obtained by a GeneCards search for the disease name. We will consider enhancing this process by also using disease aliases as search strings. We will carefully assess the potential improvement of obtained genes versus the expected introduction of noise. Moreover, we plan to scrutinize the sources that stand behind the original gene–disease associations, either by manual curation or by automatic text mining. Finally, we will use GeneCards’-rich information on genetic variations to improve the portrayal of disease-linked variations, and the recent vast enhancement of non–protein-coding RNA gene listing in GeneCards (26) to show (for example) miRNA–disease associations. Any improvement of the disease-related gene list will also positively impact annotations obtained by the GeneDecks set analysis mechanism.
Gene-independent mining. Currently, a significant portion of disease annotation is gene-based, obtained by the mechanisms of GeneCards search and GeneDecks set analysis. We will mount a major effort to introduce more direct (gene-independent) mining of disease-specific information for the relevant sections, including from external sources (e.g. publications and human phenotypes) and within MalaCards (e.g. anatomical context). This is particularly significant for the large set of diseases that have no associated genes.
Clinical trials, symptoms and phenotypes. Clinical trials will be mined for each disease before database generation, to allow for effective unification of entries representing the same trials performed at different locations. Information regarding disease symptoms will be expanded, using existing ontologies, such as the ‘Symptoms ontology’ (27) and ‘Human Phenotype Ontology’, which maps nearly all clinical descriptions in OMIM that are used more than once to an ontological structure (28). Clinical synopsis from OMIM, which represents affected body parts, tissues or systems, as well as the resultant pathophysiology, will also be linked and used in the anatomical compartments section. Future versions will contain human phenotypes from GenomeRNAi (29), recently added to GeneCards.
Related diseases. One way to associate among diseases is by looking for lexical similarity among their names. This can be done by introducing the necessary changes to our existing name unification algorithm. Such changes include adding a larger collection of terms to the list of words to be removed from the canonical form, e.g. descriptive words like ‘dominant’ and ‘autosomal’. Using such an algorithm, even broader disease types and families will be grouped together. For example, ‘hemophilia a’, ‘hemophilia a, acquired’ and ‘hemophilia a, congenital’ will be associated through the sorted canonical form ‘a hemophilia’.
Gene expression. This section will be expanded to include more normal tissues, as well as to diseased tissues. We will also seek a more direct means of relating gene expression to disease.
MalaDecks. We intend to increase MalaCards usability by implementing additional algorithms to look for shared annotations within sets of diseases, similar to what is done in GeneDecks for sets of genes (17).
Improved search. The MalaCards search will be improved and extended to allow for advance searches within user-defined fields, as well as a mechanism showing the hit context, similarly to the mini-card mechanism in GeneCards (15). Moreover, query string spell correction/dialect distinction mechanisms will be implemented.
Disease classification. This is a crucial task that has only partially and heterogeneously been tackled in the sources and books we have reviewed. We intend to supply a few classification types for each disease using an advanced tagging mechanism. Classifications can be based on grouping diseases of similar anatomical etiology, genetic/infectious/autoimmune characterization, affected organ systems and more. Rather than grouping these diseases in one hierarchical structure, we intend to tag them according to their different classification types.
Ontological mapping. Another essential feature is connecting diseases to related ontological concepts, using the MalaCards integration algorithms and other resources, like MetaMap (18). This can also complement the current use of cross-references already defined by a subset of our sources, for example, the DO, which includes cross-mapping and integration of MeSH, ICD, NCI’s thesaurus, SNOMED-CT and OMIM diseases.
Network analysis
A significant facet of the MalaCards project is its capacity to improve the understanding of disease networks. Disease network analysis has emerged as a powerful way of studying biomedical phenomena (30). Network edges may represent diverse associations between biomedical entities, such as shared genes (20), shared metabolic pathways (31), shared miRNA (32) or comorbidity (33). Analysis of disease network topology allows getting a global understanding of underlying relationships (30, 34). The revealed interactions, in turn, may unravel many unexpected links between apparently unrelated biological processes (35), as well as boost the quest for novel therapeutic strategies. In particular, it was found that related diseases (gene-based association) might arise due to dysfunction of common biological processes in the cell (34). Moreover, diseases that share genes show elevated comorbidity (36).
A comprehensive study by Barabasi et al. (20) found that genes associated with similar disorders show both higher likelihood of physical interactions among their protein products and higher expression profiling similarity for their transcripts, supporting the existence of distinct disease-specific functional modules. Moreover, the majority of disease genes are non-essential and show no tendency to encode hub proteins.
The preliminary presentation of a MalaCards-based disease network provided in this article reveals interconnection among diseases that were not known before. The edges in MalaCards are defined based on searches within MalaCards, highlighting three types of interconnections: (i) textual co-occurrence, e.g. in the ‘Summaries’ and ‘Publications’ sections, (ii) symptoms/phenotypes, and (iii) gene sharing, as manifested in the related diseases section derived from GeneDecks gene set analysis. Interestingly, we find that diseases, which are connected in the MalaCards network, are associated via three factors: etiology, common clinical features and/or clinical condition. These new connections can potentially aid in finding new candidate drugs for off-label use. It would be interesting to explore whether a similarity exists in expression profiles and protein interactions for genes associated with MalaCards-connected diseases, as in previously reported disease networks (20).
Notably, a power-law distribution is portrayed also in the more broadly disposed MalaCards-based network, which also includes non-genetic diseases, as well as edges that are derived from data other than gene sharing, similar to behavior in previously reported disease networks (20). This may imply an inherent emergent property of disease networks.
Biological discovery
A notable strength of MalaCards is its capacity to facilitate biological discovery. The association of diseases with genes, pathways and processes is a central theme of present-day research scrutiny. As an example, we describe how our ongoing research is assisted by the power of MalaCards. We are currently engaged in a collaborative study of a neurodegenerative disease, spastic paraplegia (hereditary spastic paraplegia or spastic paraparesis), characterized by progressive muscle stiffness (spasticity) and the development of paralysis of the lower limbs. We discovered a new form of the disease and identified its causative mutation in the tectonin β-propeller repeat containing 2 (TECPR2) genes (37). Our research also unveiled this as the first link of a member of the spastic paraplegia disease family to autophagy, a cell’s degradation of dysfunctional cytosolic components in the lysosome. We are currently attempting to obtain a better understanding of how aberrant autophagy may lead to disease in general and to neurodegeneration in particular. For this, we used MalaCards’ capacity to relate a search string to a large diversity of diseases, based on its multi-source disease-centric textual information.
The original connection of TECPR2 to the autophagy network was discovered in a large siRNA screen that resulted in a detailed autophagy gene interaction network (38). In this network, TECPR2 is proposed to be directly linked to the human paralogs of yeast ATG8, including the microtubule-associated protein 1 light chain 3 α and β (MAP1LC3A and MAP1LC3B). One of the diseases that came up in a MalaCards search with ‘autophag* MAP1LC3*’ is Huntington’s disease. We note that a search in PubMed for MAP1LC3* yields 280 results that need to be subjected to a further somewhat cumbersome screen for a disease relation. As the major Huntington gene, HTT (huntingtin), may play a role in microtubule-mediated transport, this MalaCards result focuses our attention on such a mechanism, not explored in the first study, as an important topic for further experimental scrutiny.
Implementation
The data collection and integration process, which runs periodically (typically every 3–5 months) to ensure ongoing access to recent updates, culminates in producing an integrated relational database (Figure 6).
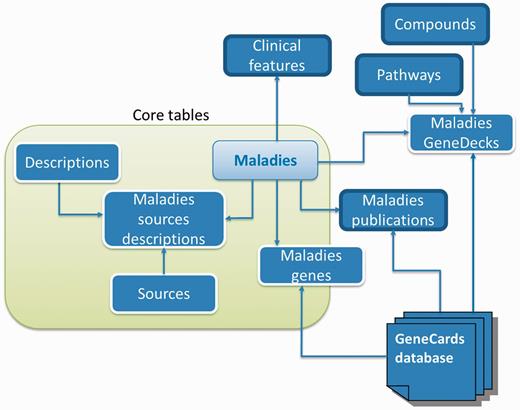
Database schema. A subset of MalaCards disease-centric relational database entities and their relationships, with associated web-card sections shown outlined in bold black.
MalaCards uses MySQL (www.mysql.org) with default MyISAM tables, PHP together with CakePHP (cakephp.org), a rapid development PHP framework with elegant MVC (model/view/controller) conventions and the Lucene search engine (http://lucene.apache.org/) powered by Solr (http://lucene.apache.org/solr/). GeneDecks’s Set Distiller server is written in Java (17). Network images were produced using the Gephi toolkit (https://gephi.org/toolkit/).
Quality assurance
Before releasing a version of MalaCards, the system undergoes a semi-automated QA process. An in-house tool verifies the integrity of the database by comparing it with that of the previous version, and it highlights inconsistencies and extreme results. The anomalies are then manually reviewed. Next, cards and their links for a sample set of diseases are manually checked by our QA professional and a medical doctor consultant. As our heuristics are still evolving, problematic disease names (e.g. ‘Interferon’ or ‘memory’) are entered into a ‘cheat list’ and removed from the system, with suggestions for improvements ticketed in our Bugzilla databases (http://www.bugzilla.org/), to be addressed in future releases.
Accessibility
The database is freely available for educational and research purposes by non-profit institutions at http://www.malacards.org. Commercial usage requires a license from LifeMap Sciences Inc. Version 1.03 launch date—6 February 2013.
Funding
LifeMap Sciences Inc., CA (USA); Crown Human Genome Center at the Weizmann Institute of Science; SysKid EU FP7 project (241544). Funding for open access charge: LifeMap Sciences Inc., CA (USA).
Conflict of interest. None declared.
Acknowledgements
The authors thank D. Warshawsky and A. Rinon for helpful discussions and ideas.
References
Author notes
Citation details: Rappaport,N., Nativ,N., Stelzer,G., et al. MalaCards: an integrated compendium for diseases and their annotation. Database (2013) Vol. 2013: article ID bat018; doi: 10.1093/database/bat018.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}