





Abstract
The CrossTope is a highly curate repository of three-dimensional structures of peptide:major histocompatibility complex (MHC) class I complexes (pMHC-I). The complexes hosted by this databank were obtained in protein databases and by large-scale in silico construction of pMHC-I structures, using a new approach developed by our group. At this moment, the database contains 182 ‘non-redundant’ pMHC-I complexes from two human and two murine alleles. A web server provides interface for database query. The user can download (i) structure coordinate files and (ii) topological and charges distribution maps images from the T-cell receptor-interacting surface of pMHC-I complexes. The retrieved structures and maps can be used to cluster similar epitopes in cross-reactivity approaches, to analyse viral escape mutations in a structural level or even to improve the immunogenicity of tumour antigens.
Database URL: http://www.crosstope.com.br
Introduction
A great challenge in immunology is the discovery of new targets to the development of vaccines against even common infectious diseases. The choice of these targets depends on the identification of elements responsible for the stimulation of immune responses. Evidence for the sharing of these elements was first provided by cross-reactivity studies (1–4). Cross-reactivity is defined by the ability of a given T-cell population to recognize different peptide:major histocompatibility complex (MHC) class I complexes (pMHC-I). This phenomenon plays an important role in antiviral cellular immunity, and there are several described cases of heterologous immunity involving completely unrelated viruses. Additionally, it has also been suggested that knowledge about the molecular features responsible for cross-reactivity can be applied to the development of a new generation of wide-spectrum viral vaccines (5).
Classical studies use epitope sequence data for selection of targets to be subsequently used in in vivo experiments. However, these data lack information about elements that are crucial for the stimulation of an appropriate immunity, such as topology and charges distribution of the molecule. These targets (peptides) are presented to the immune system in the context of MHC-I molecules, and essential structural information can be obtained from crystals of these pMHC-I complexes. These structures are determined mainly by X-ray diffraction and nuclear magnetic resonance, providing a general view of the pMHC-I surface that interacts with the T-cell receptor (TCR). Such complexes can be used for comparison between different epitopes presented by the same MHC or the same epitope presented by different MHC alleles, and they can be applied on vaccinology studies, pathogenesis of autoimmune diseases, as well as for the selection of therapeutic targets for cancer treatment. However, the number of pMHC-I structures available at the Protein Data Bank (PDB) is extremely low compared with the number of different pMHC-I complexes that can be generated by the combination of a given MHC-I allele and potential pathogen-derived immunogenic targets (thousands in a single pathogen). Until September 2012, there were ∼430 pMHC-I structures available at PDB (http://www.rcsb.org), including redundant complexes, and ∼6252 human MHC-I alleles described at the HLA (Human leucocyte antigens) Nomenclature website (http://hla.alleles.org/nomenclature/stats.html), encoding ∼4578 different proteins (allotypes). Additionally, the process of protein crystallization is highly costly and time-consuming for a large-scale prospecting study of therapeutic targets. An interesting alternative is the acquisition of such structures through in silico approaches (molecular modelling), which are considerably faster and cheaper.
There are already some crystal structures of pMHC complexes available, and there are many softwares and databases allowing reliable homology modelling of proteins, such as Modeller (6), ModBase (7), HHPred (8), Phyre (9), I-tasser (10) and so forth. However, it is important to note that pMHC modelling is not a trivial homology modelling procedure, and there are no available servers able to generate reliable models of pMHC complexes. To understand the complexity of this topic, we must divide the problem into ‘MHC modelling’ and ‘peptide modelling’. The MHC 3D structure is conserved in terms of secondary structure and global arrangement of its heavy chain domains, which allow successful modelling of different alleles, even using as template an MHC allele with important differences in the cleft conformation. For instance, our group was able to perform the cross-modelling of two different murine MHC-I alleles, H2-Db and H2-Kb, demonstrating that our modelling procedure with Modeller software was able to reproduce all aspects of the MHC-I cleft, despite the known differences in the template (11). On the other hand, there is no completely reliable template to perform epitope modelling inside the MHC cleft. ModBase (7), for instance, has tools to model small peptides, but here we are not discussing the natural folding of a given peptide. As previously discussed by our group, the conformation of the peptide inside the MHC-I cleft is not given by its amino acid sequence or its own properties, but it is rather imposed by the MHC-I cleft conformation and requirements (12). Even the very same peptide will adopt different conformations when presented by different MHC-I alleles (13); therefore, even if the same epitope is already available in a pMHC-I crystal structure, it might not be a good template for modelling.
We have considered these issues to develop a reliable technique for building pMHC-I complexes (D1-EM-D2) (12), which has been validated for human (HLA-B*27:05 and HLA-A*02:01) and murine alleles (H2-Kb and H2-Db). Some alternative in silico techniques to construct pMHC complexes have already been developed (14–16), but the question relying above all current available approaches is how reliable are these modelled structures, considering the importance of the information that they carry? In immunology, it is not enough for a model to just meet the basic stereochemical conditions of molecular modelling. For instance, disagreements in the residues that contact the TCR could compromise approaches that attempt to explain subtle differences between epitopes presented by a given MHC-I allele (17). Differences among the available methods for pMHC structure prediction can be found in a recent review published by Antunes et al. (18). In our procedure, we are (i) using a high-resolution X-ray structure from the MHC of interest, which we will call ‘MHC donor’, (ii) fitting the target epitope backbone to an allele-specific pattern conformation (previously determined) and (iii) using a reliable molecular docking software (19) to identify the best conformation for each epitope’s side chain inside the cleft. Furthermore, we also perform an energy minimization step of the entire pMHC structure—to adjust the ‘MHC donor’ cleft to this new epitope—followed by a second round of molecular docking. A general overview of this modelling process is depicted in Figure 1. The accuracy of our approach was confirmed by blind reproduction of crystal pMHC structures, presenting 8mers, 9mers and 10mers, including human and murine restricted peptides. Forty-six crystal structures were successfully reproduced with Root-mean-square deviation (RMSD) values of 1.754 ± 0.4675 Å (for all epitope atoms, not only backbone atoms) (12). Besides to provide complexes with low-RMSD values when compared with the reproduced crystal structures, our approach was also able to reproduce the molecular characteristics of the TCR-interacting surfaces (Figure 2). This approach was successfully applied to identify molecular features involved with immunogenicity variation among naturally occurring variants of an immunodominant hepatitis C virus (HCV)-derived epitope, indicating a shared pattern of charges distribution among complexes that stimulate an immune response (20). In addition, it is important to consider the difficulty to reproduce currently proposed approaches, and the limited access to the data already generated by them. Considering these issues, the CrossTope Structural Data Bank was created: the first repository of 3D structures exclusively of peptide:MHC complexes, including curated data on immunogenicity, similarity relationships and cross-reactivity.
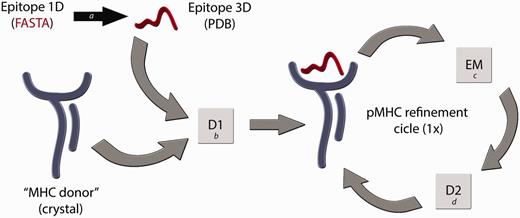
Flowchart of the D1-EM-D2 approach. The structure of a given pMHC complex not yet determined by experimental methods can be modelled through the sequential use of Docking–Energy Minimization–Docking (D1-EM-D2). The linear amino acid sequence of this peptide can be used as input for a PyMOL script (a) that generates a coordinate file (PDB) of the target epitope, fitting this backbone conformation to an allele-specific pattern. A reference crystal structure with the MHC of interest is used as an ‘MHC donor’, removing the epitope from its cleft. The ‘MHC donor’ and the ‘Epitope 3D’ are used as input for a molecular docking with Autodock Vina (b), generating a new pMHC. In this step, only the side chains of the epitope remain flexible, whereas the epitope backbone and the entire MHC structure will be kept rigid. To adjust the MHC cleft to this new epitope, an energy minimization (EM) step of the complete pMHC complex is performed with Gromacs package (c), followed by a second docking (D2) (d), which will generate the optimized final structure of the desired pMHC complex.
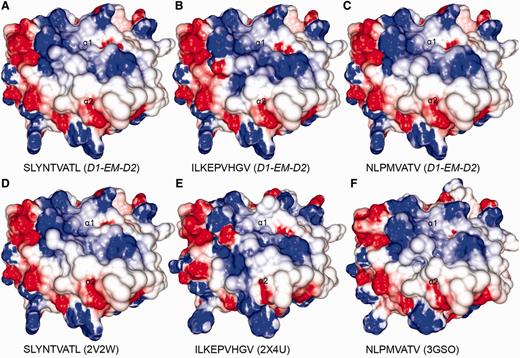
Comparison between modelled pMHCs and crystal structures. Three complexes obtained through D1-EM-D2 approach (A–C) were compared with crystal structures presenting the same epitopes in the context of the same human MHC allele (D–F). These crystal structures were included in the CrossTope DB under the IDs A0201_0095, A0201_0101 and A0201_0102, respectively. The modelled structures are similar to the reference crystal structure, especially regarding the charges distribution pattern over the TCR-interacting surface. Minor topology differences, mainly in side chain orientations, are observed because of random differences in the crystallization process. The 2V2W crystal was used as ‘MHC donor’ to build the three modelled complexes.
Structures construction and obtention, query and retrieval
CrossTope database consists of structures built through our previously commented approach and by crystallographic structures obtained from the Protein Data Bank. In the first case, new pMHC complexes are continuously included through manual curation of updated literature, searching for immunogenic epitopes that will pass through our modelling process (Figure 3A). Immunogenic epitopes included in the CrossTope database are supposed to elicit an in vitro and/or in vivo CTL immune response. Such response must have been experimentally tested. Information about the epitope immunogenicity is provided in the section ‘Epitope Information’, item ‘Immunological background’. In the same page, there is a reference that presents the tests that testify the epitope immunogenicity, as well a link to the Immune Epitope Database (Epitope ID by IEDB), where such information will also be found (in a more detailed way). This same literature revision criterion, searching for experimental evidence, was used to retrieve cross-reactive epitopes. Such information was also manually curated. In the second case, crystallographic structures are included by the search of MHC crystals also complexed with immunogenic epitopes (Figure 3B). When the search for crystals recover two structures from the same peptide:MHC complex (considered as redundant structures), only the best resolution structure is included in CrossTope database. Information regarding the inclusion of each complex is available on the main page of each complex, specifically in the item ‘Structure Type’, section ‘Complex Information’. Structure that was obtained by the modelling process is indicated as ‘Model (D1-EM-D2)’ and crystal structures as ‘Crystal’, with its respective PDB ID, which is linked to the PDB structure into the RCSB Protein Data Bank. It is important to note that the CrossTope database contains specially models, and the crystals are the minority. As previously commented, the crystals inclusion criteria follow a manual curated process, searching for structures containing immunogenic epitopes. Thus, it is possible that some crystals already available have not yet been included.
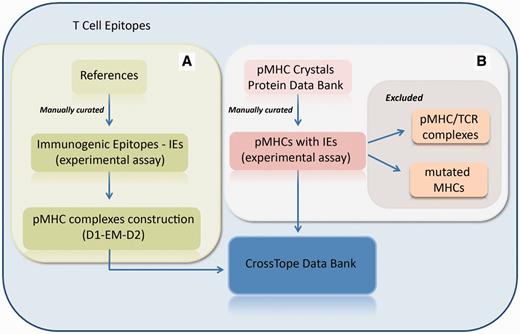
Flowchart of the inclusion of new complexes. (A) New pMHC complexes are continuously included through manual curation of updated literature, searching for immunogenic epitopes that will pass through our modelling process (D1-EM-D2). (B) Structural databanks are continuously searched for pMHCs carrying immunogenic epitopes. In this process, pMHC/TCR complexes and complexes presenting mutations on MHC α-chain are excluded. When the search recovers two structures from the same pMHC (considered as redundant structures), only the best resolution structure is included in CrossTope database.
At the time of writing CrossTope contained 182 ‘non-redundant’ structures deposited (169 models and 13 crystals) that belong to two different human alleles (HLA-A*02:01 and HLA-B*27:05) and two murine alleles (H2-Db and H2-Kb). As aforementioned, we have included only crystals with immunogenic, non-redundant epitopes, i.e. crystals of the same MHC complexed with different epitopes. Crystals presenting mutations in the MHC α-chains were also excluded. In this way, our crystal sample is inferior to the total number of pMHC-I structures available in PDB. The search for structures can be done by several ways: by MHC allele (this option also presents the image of the structural pattern adopted by the epitope for the respective allele); by peptide sequence; by source protein; and by source organism. The search returns an immunogenic target list, except when the search is done for a specific sequence of the peptide. By clicking the plus icon in a specific epitope from the list, an output containing a complete description (manually curated) of that target is generated (Figure 4): complex code, source protein and source organism, epitope position, immunological background (cross-reactivity data and immunogenicity degree presented by the epitopes) and original reference of the epitopes, as well as links to the major databases in the area (NCBI: http://www.ncbi.nlm.nih.gov/; Uniprot: http://www.uniprot.org/; and IEDB: http://www.iedb.org/). At the bottom of the page, there is a JAVA-integrated molecular viewer, JMOL (21), where it is possible to perform the initial visual inspection of the complexes. Below, it is possible to download structural coordinates in .pdb format file, as well as topology and charges distribution files (−5/+5 and −10/+10 kiloteslas) in .jpg format, which can also be viewed online at the top of the page. It is also possible to select two epitopes from the list and compare them via the ‘compare’ option. This function facilitates the comparison of the topology and charges distribution of TCR-interacting surfaces from two pMHC-I complexes, to inspect putative targets for stimulation of cross-reactivity against different epitopes (see the example later in the text).
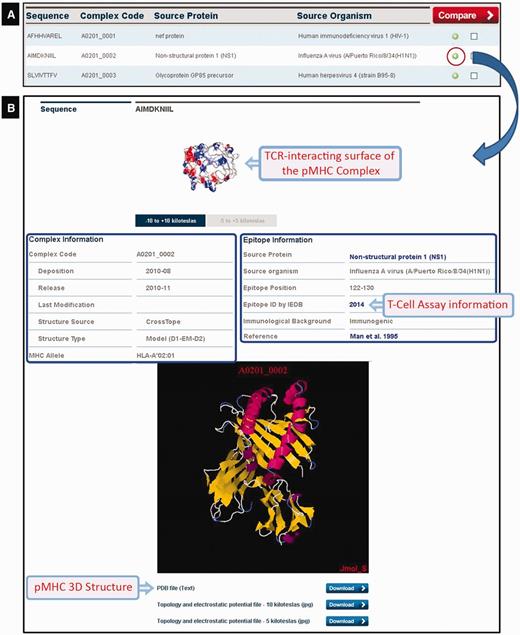
CrossTope DB output. (A) Partial list of recovered complexes in a search by MHC (HLA-A*02:01). (B) Clicking the plus icon, the user can access information regarding both the epitope (manually curated) and the pMHC complex. Links to IEDB and Uniprot are also provided, as well to download coordinate and charges distribution files.
Example—use of multivariate statistical methods for structural virtual screening of cross-reactive targets
A well-known case of cross-recognition involving the epitopes IV-M158–66 (GILGFVFTL) and HIV-GAG77–85 (SLYNTIAVL) was investigated through the surface analysis, using the ‘compare’ option. The visual inspection revealed a striking similarity between them, evidencing the reliability of this kind of investigation. Here, we extended a previous study (20) and analysed 60 unrelated pMHC-I complexes presenting virus-derived peptides, in the context of the most frequent human MHC allele (HLA-A*02:01). These complexes, 5 crystal structures and 55 in silico predicted structures, were obtained from the CrossTope. Images of the TCR-interacting surface of these complexes, presenting the electrostatic potential distribution (Supplementary Figure S1), were used to extract the RGB (Red-Green-Blue) colour histograms of seven selected regions. These regions were selected considering the spots of variation in charges distribution over the pMHC-I surface, and they are placed within an area corresponding to the already described footprints of public TCRs (22). Values of mean and standard deviation of the three RGB components, for each one of these selected areas, were used as input for multivariate statistical methods, to predict possible targets of cross-reactivity.
Our dataset included some peptides with already known cross-reactivity, and the hierarchical cluster analysis (HCA) results were in agreement with the experimental background (Supplementary Figure S2). For instance, we included 10 variants of the wild-type immunodominant epitope HCV-NS31073 (CV/INGVCWTV). The wild-type and all the cross-reactive variants (genotypes 4, 5 and 6) fall in the same group, whereas the non–cross-reactive variant (genotype 3) falls in a completely unrelated group. Both complexes containing the epitopes IV-M158–66 (GILGFVFTL) and HIV-GAG77–85 (SLYNTVATL) grouped together. Interestingly, this cluster also contained the cross-reactive variants of HCV-NS31073 epitope. Cross-reactivity between this HCV immunodominant epitope and the HIV-GAG77–85 peptide has not been described so far. There is yet other two complexes included in the same cluster, presenting the ‘LLWTLVVLL’ and the ‘NLVPMVATV’ peptides, from the human herpes virus 4 (LMP2329) and 5 (pp65485), respectively. It is important to note that the former peptide does not share even a single amino acid with the target peptide (CV/INGVCWTV) and, nevertheless, presented almost the same topology and charges distribution when presented in the context of HLA-A*02:01. Nevertheless, this putative cross-reactivity remains to be confirmed.
Discussion
The CrossTope Structural Data Bank opens a way for the exploration of an additional level of complexity of immunogenic epitopes, the comparison at the molecular level, hitherto confined to analysis of scarce pMHC-I complexes. For now, our approach is restricted to MHC alleles containing a sufficient number of different epitopes in pMHC-I crystals, so that the allele-specific structural pattern of them could be inferred (12). The alleles already available include two murine MHCs largely used for in vitro/in vivo assays of immunogenicity and cross-reactivity, and also two key human MHCs. HLA-A*02:01 is one of the most frequent human MHC alleles (http://www.allelefrequencies.net/), and HLA-B*27:05 has important roles in autoimmunity (23,24) and also in viral control of HCV and HIV (25–27). Additionally, our expectation is that we can perform the continuous inclusion of new complexes (including new MHC alleles) and the development of automated tools (clustering cross-reactive targets).
The CrossTope Database focuses on Cytotoxic T Lymphocyte (CTL) immune response. Thus, only pMHC-I alleles will be included. Moreover, epitopes restricted to MHC-II present a variable number of amino acids, and the structural patterns are not as conspicuous as in MHC-I epitopes. This could be explained by the differential nature (feature) of the MHC-I cleft, which presents closed extremities, forcing in a more explicit manner the epitopes to adopt more stringent structural patterns. Moreover, for MHC-II epitopes, we would need to define which core region would be located inside the cleft, and this is more difficult to define, even for sequence predictors.
Considering that these pMHC-I complexes are the putative carriers of the immunogenic signals in cytotoxic stimulation, and that structural features of the pMHC complexes, especially regarding to charges distribution over the TCR-interacting surface, are key elements for cross-reactivity and heterologous immunity the CrossTope Database was developed to give support to researchers interested in exploring such elements.
In this context, CrossTope provides images of the charges distribution over the TCR-interacting surface of each pMHC-I available in the databank for cross-reactivity prediction (as in the example provided earlier in the text). These images can be compared on line or downloaded for further analysis. We choose two different colouring spectra to represent the electrostatic potential (−5 to +5 kT and −10 to +10 kT), which will be depicted as a gradient from dark red (negative charges) to dark blue (positive charges). The PDB file for each complex is also provided, allowing the users to generate their own charges distribution file through the GRASP2 program (28), as well as to perform other structural analysis. It is important to note that the most important regions for predicting cross-reactivity in one subset might vary according to the MHC allele and even the T-cell population that is being considered. Therefore, here we just provided one example of how these surface images can be used to make predictions about cross-reactivity. Other researchers might want to define their own selected regions for analysis. In any case, the regions contacted by the TCR will be represented within the TCR-interacting surface, which are represented in the images available in our database.
Our ultimate goal is to provide a platform that allows scientists to perform the prospection of new cross-reactive targets, or even to identify the molecular basis for triggering an adequate immune response, envisaging a new generation of vaccines.
Funding
CNPq, CAPES (Process No 23038.035722/2008-19) and a grant from Bill & Melinda Gates Foundation through the Grand Challenges Exploration Initiative (Grant ID 53049). Funding for open access charge: Programa de Pós-Graduação em Genética e Biologia Molecular (PPGBM/UFRGS).
Conflict of interest. None declared.
Acknowledgements
This research was performed with resources from the Centro Nacional de Supercomputação da Universidade Federal do Rio Grande do Sul (CESUP/UFRGS). The authors thank Jader Peres da Silva and Francis Maria Báo Zambra for their inestimable help in the construction of pMHC-I complexes.
References
Author notes
Citation details: Sinigaglia,M., Antunes,D.A., Rigo,M.M. et al. CrossTope: a curate repository of three-dimensional structures of immunogenic peptide: MHC complexes. Database (2013) Vol. 2013: article id bat002; doi: 10.1093/database/bat002

{kind=link}
{kind=link}
{kind=link}
{kind=link}