





Abstract
We recently developed a user-friendly web-based application called bc-GenExMiner (http://bcgenex.centregauducheau.fr), which offered the possibility to evaluate prognostic informativity of genes in breast cancer by means of a ‘prognostic module’. In this study, we develop a new module called ‘correlation module’, which includes three kinds of gene expression correlation analyses. The first one computes correlation coefficient between 2 or more (up to 10) chosen genes. The second one produces two lists of genes that are most correlated (positively and negatively) to a ‘tested’ gene. A gene ontology (GO) mining function is also proposed to explore GO ‘biological process’, ‘molecular function’ and ‘cellular component’ terms enrichment for the output lists of most correlated genes. The third one explores gene expression correlation between the 15 telomeric and 15 centromeric genes surrounding a ‘tested’ gene. These correlation analyses can be performed in different groups of patients: all patients (without any subtyping), in molecular subtypes (basal-like, HER2+, luminal A and luminal B) and according to oestrogen receptor status. Validation tests based on published data showed that these automatized analyses lead to results consistent with studies’ conclusions. In brief, this new module has been developed to help basic researchers explore molecular mechanisms of breast cancer.
Database URL:http://bcgenex.centregauducheau.fr
Introduction
The increasing amount of genomic data represents a new resource for fundamental and translational research, but it is limited in its use due to complexity and heterogeneity of the different studies; therefore, in its raw form, it still remains underexploited. To fully take benefit from this resource, bioinformatics processes, which preserve biological sense caught in annotated genomic data, have to be applied before developing automatized mining functionalities, e.g. biostatistics’ analyses. We have recently developed a user-friendly web-based application called bc-GenExMiner, which offered the possibility to evaluate prognostic informativity of genes in breast cancer by means of a ‘prognostic module’ including three functionalities (1). Statistical analyses were based on genomic data and corresponding bioclinical annotations of 21 studies. In this study, numerous biological tests demonstrated that biological sense contained in breast cancer tumours was preserved despite data origin diversity and bioinformatics process complexity, even when data were merged in new cohorts. This development confirmed our opinion that such data and automatized statistical tests could actually help researchers to find new prognostic markers or therapeutic targets. Hence, we have developed a new module called ‘correlation module’, which includes three kinds of gene expression correlation analyses. The first one computes correlation coefficient between 2 or more (up to 10) chosen genes. The second one produces two lists of genes that are most correlated (positively and negatively) to a ‘tested’ gene. A gene ontology (GO) mining function is also proposed to explore GO ‘biological process’, ‘molecular function’ and ‘cellular component’ terms enrichment for the output lists of most correlated genes (2). The third one explores gene expression correlation between a ‘tested’ gene and each of the 15 DNA 5’- and 15 3’-closest genes surrounding it. The aim of the last functionality is to identify DNA continuous clusters of correlated co-expressed genes, which can be linked to genomic anomalies, including chromosomal aberrations [e.g., copy number alterations (CNAs)]. These correlation analyses can be performed in different groups of patients: all patients (without any subtyping), in molecular subtypes (basal-like, HER2+, luminal A and luminal B) and according to oestrogen receptor (ER) status (3, 4). The interest of testing these subgroups of patients is based on the fact that CNAs differentially affect molecular subtypes (4–6). Validation tests based on published data showed that these automatized analyses lead to results consistent with studies’ conclusions. In brief, this new module has been developed to help basic researchers explore molecular mechanisms of breast cancer.
Materials and methods
System architecture and database content
System implementation, data selection and data pre-processing are fully described elsewhere (1).
Briefly, bc-GenExMiner is powered by Apache with a MySQL relational database storage. Web interfaces are written in PHP v5 and JavaScript. Statistical analyses are performed with R software. Datasets included in the database were publicly available (Gene Expression Omnibus, ArrayExpress, Stanford microarray database and also on author’s individual web pages). Non-Affymetrix platform data were ratio-normalized, and Affymetrix raw CEL data were MAS5-normalized. Data were then log2-transformed. Finally, to merge data of all studies and create pooled cohorts, data were converted to a common scale (median = 0 and standard deviation = 1).
bc-GenExMiner functionalities
A flowchart details purpose of analyses (Figure 1).
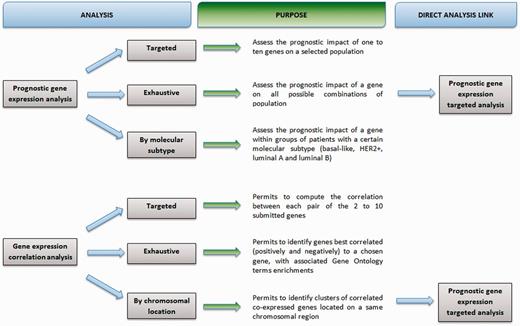
bc-GenExMiner 3.0 flowchart.
ER status genomic determination
In cohorts GSE1456, GSE3143 and GSE11121, ER status determined by immunohistochemistry was not available. For these Affymetrix® cohorts, ER status was determined based on 205225_at Affymetrix® probes (U133 array; cohorts GSE1456 and GSE11121) or on the median values of Affymetrix® probes representing ESR1 (U95 array; cohort GSE3143) using a two-component Gaussian mixture distribution model (3).
Gene expression correlation analyses
Gene correlation targeted analysis
In a gene correlation analysis, the user chooses 2 to 10 different genes, and all possible pairwise Pearson’s correlation coefficients are computed, along with the associated P-value, on merged datasets based on seven different populations: all patients pooled together or subsets of patients by ER status (two subsets) and molecular subtype (four subsets). Molecular subtype annotation was performed by means of six molecular subtype predictors (MSPs) and lead to robust molecular subtype predictor classification (RMSPC), which only includes patients with concordant molecular subtype assignment for the 6 MSPs (1). Results are displayed in a correlation map, i.e. a table where each cell corresponds to a pairwise correlation and is coloured according to the correlation coefficient value, from dark blue (coefficient = −1) to dark red (coefficient = 1). Correlation plots are also drawn to illustrate each pairwise correlation.
Gene correlation exhaustive analysis
A gene correlation exhaustive analysis permits to know the genes that are best correlated, positively and negatively, to a given gene, chosen by the user: Pearson’s correlation coefficient is computed between the chosen gene and all other genes that are present in bc-GenExMiner database. Analyses are computed on merged datasets and are based on five different sets of patients: all patients, RMSPC basal-like patients, RMSPC HER2+ patients, RMSPC luminal A patients and RMSPC luminal B patients. Genes with coefficient above 0.40 in absolute value and with significant associated P-value (<0.05) are retained.
As a complement, GO terms directly linked to genes are screened to identify those underrepresented or overrepresented in the lists of genes that are most positively correlated to the chosen gene (including itself), most negatively correlated to the chosen gene or in the union of the two previous lists.
Results are presented in two tables: one for the positive correlations and one for the negative correlations. For each table, only the 50 best genes are displayed; the full lists can be further downloaded. A GO button permits to visualize GO ‘biological process’, ‘molecular function’ and ‘cellular component’ terms significantly associated with the gene lists. GO analyses are based on the complete lists of genes, which are significantly correlated (r ≥ 0.4; P < 0.05) with the tested gene.
Gene ontology analysis
This functionality is based on the mining of the three GO trees (biological process, molecular function and cellular component) (2). A GO analysis finds GO terms that are significantly linked to a given list of genes (‘target list’). For each term of each of the GO trees, comparison is done between the number of occurrences of this term in the ‘target list’, i.e. the number of times this term is directly linked to a gene, and the number of occurrences of this term in the ‘gene universe’ (all of the genes that are expressed in the database) by means of Fisher’s exact test (P < 0.01 is considered significant for this analysis). GO terms database was downloaded from geneontology.org and will be updated every 6 months as gene annotations.
Gene correlation analysis by chromosomal location
DNA location data were computed with information extracted from The Ensembl Genome Database (http://www.ensembl.org), NCBI Map Viewer (http://www.ncbi.nlm.nih.gov/mapview/) and NCBI Unigene (http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene) websites. Only gene position (start and end) and DNA strand (positive or negative) were kept in our final list of chromosomal locations.
In a gene correlation analysis by chromosomal location, the user chooses one gene, and Pearson’s correlation coefficients, with associated P-values, are computed on merged datasets between this gene and genes continuously located around it on the same chromosome (15 centromeric genes and ≤15 telomeric genes), based on all patients or on different subsets depending on the ER status or on the molecular subtype. Results are displayed in a table giving the gene location and correlation details for each of the seven different populations (all patients, by ER status, by molecular subtype). Correlation plots are also performed.
Targeted correlation analysis (TCA; ‘TCA’ button), which here aims at evaluating the robustness of clusters, is proposed: Correlation analyses are automatically computed between all possible pairs of genes that compose a selected cluster.
Biological validation
Targeted analysis
MKI67, AURKA and UBE2C: We focused on proliferation genes because of the importance of this process on breast cancer prognosis (7). Genomic studies demonstrated the existence of a proliferation cluster containing numerous correlated genes. We chose these three genes because they are known to play an active role in proliferation process in breast cancer: MKI67 is coding for KI67 protein, which is routinely explored by means of immunohistochemistry, AURKA is considered as the proliferation prototypic gene and UBE2C bears a high prognostic informativity (8, 9).
ESR1, GATA3, FOXA1 and XBP1: Numerous studies, notably based on microarrays, have shown that expressions of GATA3, FOXA1 and XBP1 were strongly correlated to that of ESR1 (10).
TNFAIP1/POLDIP2, RAF1/MKRN2 and TBCB/POLR2I: The examples of TNFAIP1/POLDIP2, RAF1/MKRN2 and TBCB/POLR2I are of particular interest because these couples of genes demonstrated co-regulatory pattern in breast cancer tumours, are located at the same locus and are organized in sense–antisense architecture on the opposite DNA strands of chromosome 17, 3 and 19, respectively (11).
Exhaustive analysis
AURKA: We chose AURKA, which is known to be the prototypic gene of proliferation process. After exhaustive gene expression correlation analysis, we verified that the 72 genes included in proliferation gene expression signature were present in AURKA’s most correlated genes list (12).
ESR1: ESR1, one of the most important genes in breast cancer physiopathology, was tested.
FTL: We recently validated ferritin light chain (FTL) as a breast cancer prognostic marker (13). In this study, we demonstrated by means of double immunofluorescence that FTL was located in tumour-associated macrophages (TAMs) harbouring an M2-phenotype (CD163-positive TAM).
Gene ontology analysis
Following gene correlation exhaustive analysis, GO biological process enrichment analyses for AURKA, ESR1 and FTL were computed to explore biological process annotations of lists of most correlated genes.
By chromosomal location
This kind of analysis pinpoints continuous clusters of co-expressed genes and permits to visualize their chromosomal organization. For all following tests, TCA was performed to verify the robustness of clusters.
An in silico study conducted by Buness et al. (14) identified 32 series of 20 characteristic overexpressed genes for amplified chromosomal regions. Six hundred and forty genes were tested for all patients and molecular subtypes. We considered as positive results continuous clusters of correlated co-expressed genes composed of at least three genes with r ≥ 0.30 and P ≤ 0.05. To quantify the global relationship among a cluster of co-expressed genes, eigenvalues of the correlation matrix were computed, and the ratio of the largest one to the sum of all eigenvalues multiply by 100 was taken (i.e. it comes to perform a principal component analysis based on the correlation matrix and take the percentage of variance explained by the first principal component) (Supplementary Method). This value is called multicorrelation score (MCS).
ESR1: A recent study showed that ESR1 was co-expressed with closely adjacent genes at 6q25.1 (15).
8p11-12 amplicon: At 8p11-12, one of the most frequently amplified regions in breast cancer (10–15% of cases), Bernard-Pierrot et al. identified five genes (LSM1, BAG4, DDHD2, PPAPDC1B and WHSC1L1) as consistently overexpressed due to an increased gene copy number (16, 17).
Chromosome 17: Chromosome 17 is highly amplified in breast cancer, especially in HER2+ molecular subtype. Its amplified regions permit to distinguish HER2+ and luminal A (5, 18). We chose three genes in different regions of chromosome 17 to test bc-GenExMiner analyses: TRAF4 at 17q11-q12, MED24 at 17q21.1 and GGA3 at 17q25.1 (14).
ER status: Numerous studies showed that CNA varied among breast tumours with different ER status (4, 5, 19–21). A list of 59 genes, which demonstrated different expression profile according to ER status in gained regions, was tested to screen for DNA continuous clusters of correlated co-expressed genes (20). We chose the following criterion: at least two DNA continuous genes with correlated gene-expressions, to define clusters of correlated co-expressed genes. MCS was used to compare clusters of co-expressed genes.
Differential gene expression according to ER status
As a complement of correlation analyses by chromosomal location within homogeneous ER status groups, expression of genes according to ER status was also studied by means of Mann–Whitney test.
Remark: As ESR1, ERBB2 and AURKA are the three genes most implicated in the molecular subtypes determination (ESR1, ERBB2 and AURKA play indeed a very important role in the determination of the molecular subtypes whatever the MSP chosen; in particular, one of the six predictors involved in the RMSPC only takes those three genes into account to determine a patient’s molecular subtype), they were not tested within each of those subtypes to avoid bias in the analysis results due to the dependency between molecular subtype determination and gene expression.
Results
Biological validation
Correlation analyses
Targeted: MKI67, AURKA and UBE2C demonstrated significant correlations in ‘all patients’: r = 0.74 for AURKA/UBE2C (n = 2928), 0.64 for AURKA/MKI67 (n = 2928) and 0.58 MKI67/UBE2C (n = 3160) with P < 10−4.
ESR1, GATA3, FOXA1 and XBP1: As suspected, bc-GenExMiner analysis showed that all these genes were correlated, with correlation coefficients ranging from 0.51 to 0.73 (Table 1).
Targeted correlation analysis of ESR1, GATA3, FOXA1 and XBP1 (P < 10−4)
| GATA3 | FOXA1 | XBP1 | |
|---|---|---|---|
| ESR1 | r = 0.56 (n = 3414) | r = 0.52 (n = 3315) | r = 0.51 (n = 3404) |
| GATA3 | — | r = 0.73 (n = 3315) | r = 0.60 (n = 3404) |
| FOXA1 | — | — | r = 0.66 (n = 3315) |
As expected, TNFAIP1/POLDIP2, RAF1/MKRN2 and TBCB/POLR2I correlation analyses showed correlated co-expressions for the three couples of genes (r = 0.51, 0.44 and 0.56, respectively, with P < 10−4).
Exhaustive: Summary of results from exhaustive correlation analyses for AURKA, ESR1 and FTL is displayed in Table 2.
Results of gene correlation exhaustive analysis and GO enrichment analysis for the biological process tree for AURKA, ESR1 and FTL, ‘all patients’ population
| Tested genes | No. positively correlated genesa | No. genes with GO biological process annotationsa | No. GO enrichment terms (P < 0.01) |
|---|---|---|---|
| AURKA | 590 | 365 | 121 |
| ESR1 | 487 | 215 | 38 |
| FTL | 281 | 172 | 78 |
aIncludes reference gene.
AURKA: Each gene belonging to the 72-gene expression proliferative signature was present in AURKA ‘correlation exhaustive analysis’ output, which meant each correlation coefficient was superior to 0.40 and significant (actually, all P-values were <10−4).
FTL: Following one of our recent work, FTL and CD163 gene expression were checked (13). FTL correlation exhaustive analysis output showed that CD163 gene was correlated with FTL gene in breast cancer groups of patients with or without molecular subtyping (r = 0.44–0.54). In a cohort of 2773 patients, CD163 was the 15th most correlated with FTL gene (Table 3). Rank varied according to molecular subtype.
Summary results of FTL gene correlation exhaustive analysis, with focus on CD163
| Breast cancer patients | No. of patients | No. FTL positively correlated genes (r > 0.4 and P < 0.05) | Rank of CD163 | CD163/FTL r (P < 0.001) |
|---|---|---|---|---|
| All patients | 2773 | 280 | 15 | 0.54 |
| Basal-like | 274 | 107 | 38 | 0.49 |
| HER2+ | 76 | 569 | 134 | 0.54 |
| Luminal A | 194 | 190 | 118 | 0.44 |
| Luminal B | 53 | 286 | 170 | 0.44 |
Gene ontology analysis: The results of GO biological process enrichment term analyses for AURKA, ESR1 and FTL are displayed in Table 2.
AURKA: As suspected, GO enrichment for biological process of AURKA’s most correlated genes essentially pointed out proliferation via the following terms: ‘mitosis’, ‘cell division’, ‘cell cycle’ and ‘DNA replication’.
ESR1: Mammary development, represented by AR, PGR, ESR1 and CCND1 genes, and oestrogen pathway, represented by EGLN2, RARA, ESR1, GATA3, BCL2, CRIPACK, CCND1, FOXA1 and AR genes, appeared in the best classified biological process terms. Prostate and male gonad development also appeared in the top of this classification because genes involved in these processes also play a major role in mammary molecular physiology (FOXA1, AR and IGF1R for ‘prostate gland epithelium morphogenesis’, BBS2 and UBE2B for ‘sperm axoneme assembly’ and ESR1, GATA3, BCL2, AR and PATZ1 for ‘male gonad development’).
FTL: GO enrichment for biological process of FTL’s most correlated genes pointed out immune response, immune cells (T and B cells, neutrophils and macrophages), immune processes and inflammatory response (Figure 2). ‘Positive regulation of macrophage chemotaxis’ included C5AR1, C3AR1 and RARRES2 genes.
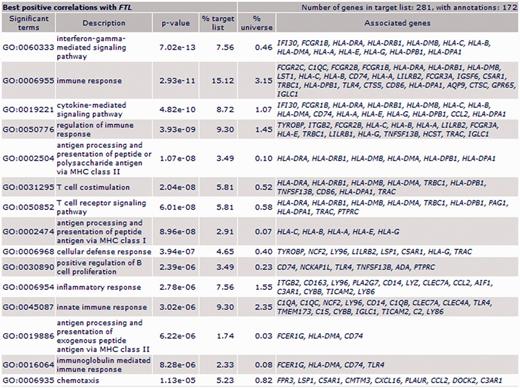
Biological process ontology enrichment results for FTL most positively correlated genes in all patients population (first 15 GO biological process terms).
By chromosomal location: On the basis of Buness data, we always found continuous clusters of correlated co-expressed genes for all 32 chromosomal regions (Supplementary Table 1). The largest cluster, located at 17q24-q25 cytoband, was composed of 12 genes (CDR2L, ICT1, ATP5H, KCTD2, SLC16A5, ARMC7, NT5C, HN1, SUMO2, NUP85, GGA3 and MRPS7) for HER2 molecular subtype. Other large clusters were identified in 17q cytobands for HER2 molecular subtype (17q12-q21, 17q11-q12 and 17q21-q23). Consistent with numerous studies, 17q-clusters were rarely found in luminal A molecular subtype.
ESR1: Among the 30 genes selected in the close vicinity of ESR1 (PCMT1 - LRP11 - RAET1E - ULBP2 - ULBP1 - ULBP3 - PPP1R14C - IYD - PLEKHG1 - MTHFD1L - AKAP12 - ZBTB2 RMND1 - C6ORF211 - C6ORF97 - [ESR1] - SYNE1 - MYCT1 – VIP - FBXO5 - MTRF1L - RGS17 - OPRM1 - IPCEF1 - CNKSR3 - RBM16 - TIAM2 - CLDN20 - TFB1M - NOX3 - ARID1B), only 3 genes, all located in a continuous amplicon containing ER gene at 6q25.1, presented a good correlation with ESR1 in all patients with breast cancer (without molecular subtyping): C6orf211, C6orf97 and RMND1 (C6orf96), even when correlation targeted analysis including these 4 genes was performed. Furthermore, level of expression of these genes according to ER status, based on bcGenExMiner data, showed that all of them were overexpressed in ER+ tumours (Supplementary Figure 1). bcGenExMiner results are concordant with Dunbier’s ones (15) (Figure 3).
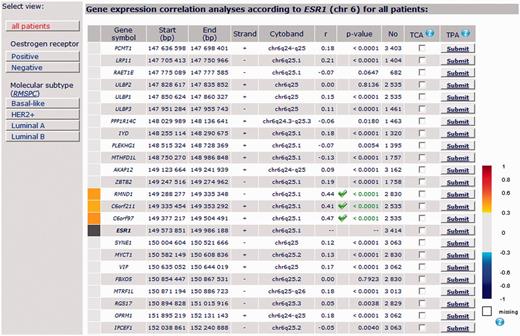
Detailed results of ESR1 gene expression correlation analysis by chromosomal location for all patients.
8p11-12 amplicon: Among the 30 genes continuously located around LSM1 gene (DUSP26 - C8ORF41 - UNC5D - KCNU1 - ZNF703 - ERLIN2 - PROSC - GPR124 - BRF2 - RAB11FIP1 - GOT1L1 - ADRB3 - EIF4EBP1 - ASH2L - STAR - [LSM1] - BAG4 - DDHD2 - PPAPDC1B - WHSC1L1 - FKSG2 - LETM2 - FGFR1 - TACC1 - PLEKHA2 - HTRA4 - TM2D2 - ADAM9 - ADAM32 - ADAM5P - ADAM3A), bc-GenExMiner correlation analysis showed a robust continuous cluster of co-expressed genes composed of LSM1, BAG4, DDHD2, PPAPDC1B and WHSC1L1 in ‘all patients’ group (Figure 4). This result was in agreement with Bernard-Pierrot’s one (16). Robustness of this cluster was demonstrated by performing a TCA with these five genes (Table 4). This cluster was also found in basal-like, HER2+ and luminal B patients. Best MCS (87.7) was obtained in luminal B patients, who display more frequent high-level DNA amplification, especially in chromosome 8 region, than other molecular subtypes (MCS = 63.1 in basal-like and 71.5 in HER2) (5) (Table 4). This last result strengthened the idea that genomic instability, here indirectly materialized by cluster of co-expressed genes, may be specific of molecular subtypes and that bc-GenExMiner may help researchers to find such genomic anomalies.
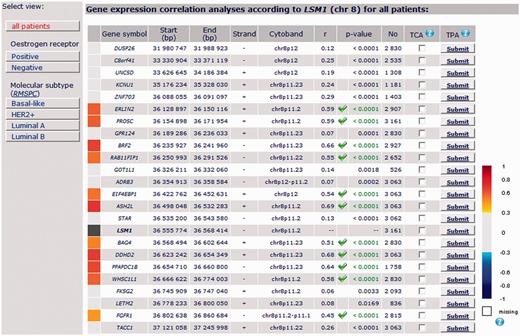
Detailed results of LSM1 gene expression correlation analysis by chromosomal location for all patients.
Targeted correlation analysis of LSM1, BAG4, DDHD2, PPAPDC1B and WHSC1L1 cluster at 8p11-12 in all breast cancer patients and within molecular subtypes
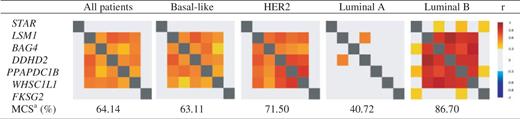 |
aMulticorrelation score for LSM1 - BAG4 - DDHD2 - PPAPDC1B - WHSC1L1 cluster.
Chromosome 17:TRAF4, MED24 and GGA3 are included in clusters of correlated co-expressed genes, composed of 8, 5 and 13 genes, respectively (Table 5). Correlation robustness was tested by means of TCA. These clusters differentiate HER2+ from luminal A subtype, which did not display the same expression profile. In this molecular subtype, there is no correlation between the genes of the above-mentioned clusters.
Robust and continuous correlated co-expressed gene clusters of chromosome 17
| Tested gene | Cytoband | Genes of the cluster | Length of non-amplified DNA region covered by the cluster (bp) |
|---|---|---|---|
| TRAF4 | 17q11-q12 | TLCD1, NEK8, TRAF4, C17orf63, ERAL1, FLOT2, DHRS13, PHF12 | 226 406 |
| MED24 | 17q21.1 | MED24, THRA, NR1D1, MSL1, CASC3 | 152 273 |
| GGA3 | 17q25.1 | C17orf28, CDR2L, ICT1, ATP5H, KCTD2, SLC16A5, ARMC7, NT5C, HN1, SUMO2, NUP85, GGA3, MRPS7 | 315 466 |
ER status: Of the 59 tested genes, 21 belonged to continuous clusters of correlated co-expressed genes specific of ER− status and 4 to amplicons of discontinuous correlated co-expressed genes (Supplementary Table 2). No ER− specificity was found for 34 of these genes. In this group, 24 were not linked to ER− or ER+ status while 10 showed ER+ specificity, the last ones are called discordant cases. In discordant cases, seven showed a basal-like specificity or tendency; these contradictory results question the robustness of ER+ specificity in most of these cases. By means of bc-GenExMiner gene expression correlation analysis by chromosomal location, we found a majority of clusters of correlated co-expressed genes linked to ER− status. Discordant results might be explained by a bias in the study by Han et al. due to low number of cases compared to those used in our analyses.
Discussion
Gene expression correlation analysis permits to explore mathematical relation between two genes; precisely, the strength of the positive or negative linear link. But one must not conclude that correlated genes are involved in the same causal chain of events, even if it might happen sometimes. Causal chain involves correlated parameters, but the reverse is untrue. We have to remind that correlation is different from causal determinism. Causal determinism means a physical link, which is time dependent as defined by the causal sequence: A cause produces an effect. In molecular biology, correlation between gene expressions might more likely deal with co-regulation process. Correlation may be used to detect causal connections, which need further and rigorous experimental investigations to be proved.
Gene expression TCA permits to explore link between pairs of genes in an intuitive manner. Therefore, the user can easily and rapidly test its intuition or benchmark results.
Gene correlation exhaustive analysis extracts genes that are correlated with the gene of interest in a non-intuitive, i.e. screening, manner. Computation involves all genes included in bc-GenExMiner genomic database (n = 20306). Non-intuitive and automatized interpretation by means of GO terms enrichment is proposed to help researchers to give a biological sense to these data.
Gene correlation analysis by chromosomal location has been developed to identify DNA continuous clusters of correlated co-expressed genes in a cohort composed of all patients or on different molecular subtype breast cancer patients, and so to evaluate whether these clusters are specific of molecular subtypes. Co-expression may be due to co-regulation process, which can be linked to DNA amplification, i.e. DNA CNA. Previous studies demonstrated that highly amplified genes (44–62%) showed moderately or highly elevated expression (22, 23). In the opposite way, in 10.5 to 12%, overexpression was directly attributable to variation in gene copy number. DNA amplification may involve one gene or several genes belonging to a same locus. Our hypothesis was that amplified or deleted genes might be co-expressed, i.e. overexpressed or not expressed, due to these genomic mutations and that our analysis might complete DNA CNA screening studies by means of comparative genomic hybridization or single-nucleotide polymorphism arrays.
Funding
Funding for open access charge: Our institution's (ICO) research budget.
Conflict of interest. None declared.
References
Author notes
Citation details: Jézéquel,P., Frénel,J.-S., Campion,L., et al. bc-GenExMiner 3.0: new mining module computes breast cancer gene-expression correlation analyses. Database (2012) Vol. 2012: article ID bas060; doi:10.1093/database/bas060.

{kind=link}
{kind=link}
{kind=link}
{kind=link}