





Abstract
As part of a communitywide effort for evaluating text mining and information extraction systems applied to the biomedical domain, BioC is focused on the goal of interoperability, currently a major barrier to wide-scale adoption of text mining tools. BioC is a simple XML format, specified by DTD, for exchanging data for biomedical natural language processing. With initial implementations in C++ and Java, BioC provides libraries of code for reading and writing BioC text documents and annotations. We extend BioC to Perl, Python, Go and Ruby. We used SWIG to extend the C++ implementation for Perl and one Python implementation. A second Python implementation and the Ruby implementation use native data structures and libraries. BioC is also implemented in the Google language Go. BioC modules are functional in all of these languages, which can facilitate text mining tasks. BioC implementations are freely available through the BioC site: http://bioc.sourceforge.net .
Database URL:http://bioc.sourceforge.net/
Introduction
The BioCreative Workshops provide a forum for text mining, computational linguistics and natural language processing researchers to build, adapt and/or integrate information extraction systems that address biologically meaningful tasks and that provide results of practical relevance. To ensure satisfactory community assessment and method comparison, and to promote scientific progress, it is necessary to establish common standards and shared criteria that enable comparison and integration of different approaches. BioC ( 1 ) has been gaining momentum as a solution to the interoperability challenge—an interchange format for biomedical natural language processing tools. Expressed in a simple XML format, specified by DTD, text documents and related data annotations can be shared easily between different text mining and information extraction systems applied to the biomedical domain. Moreover, all tools that communicate with this shared format can potentially be combined as parts of larger, more complicated systems.
Considering the variety of computational tools used by the biomedical text mining community, successful interoperability requires uniform BioC support across various programming language environments. The first releases of BioC code ( 1 ) were implemented in C++ and Java, two mainstream programming languages that provide a solid foundation for defining BioC functionality. However, scripting languages such as Perl, Python and Ruby have become popular in the communities of bioinformatics and natural language processing for their ease of use with reasonable performance. For example, BioPerl ( http://www.bioperl.org ) and Biopython ( http://biopython.org/ ) are widely adopted for the tasks of parsing BLAST output and querying the GENBANK database ( 2 , 3 ). For biomedical natural language processing tasks, a variety of NLP packages [CPAN-NLP ( http://cpan.org ), NLTK ( http://nltk.org )] have been developed with Perl and Python. For Ruby, BioRuby ( 4 ) and BioGems ( 5 ) are useful libraries for bioinformatics and biomedical applications. Go ( http://golang.org/ ), a newly emerging language from Google, is also receiving attention with biogo ( https://code.google.com/p/biogo/ ) targeted at computationally intensive Bioinformatics tasks.
Therefore, it is important to make BioC functionality easily accessible for applications that have been developed with these languages to take advantage of the aforementioned tools. With the support of BioC modules, both existent and future applications will be able to conveniently extract information from BioC files, process the information and write the output in BioC format. This facilitates efficient code reuse and uniform data sharing.
To efficiently deliver behavior identical to C++ in a wide range of programming languages, we use the interface generator SWIG ( http://www.swig.org ) to extend the C++ BioC implementation to Perl and Python. The Perl BioC extension is thus far the only option to support BioC in Perl. By supporting full BioC functionality faithfully, the integration of the C++ BioC implementation and target languages enables fast and flexible prototyping while relying on the low-level C++ code to duplicate the behavior of C++ applications. Directly implementing BioC in other programming languages is another solution. Although careful testing is required, complete compatibility can be easily achieved. It enables BioC users to seamlessly integrate BioC modules with their data processing. This is the approach taken in our Go and Ruby implementations and a second Python version.
In this article, we introduce the effort of our team to enable BioC users to take advantage of BioC utilities in Perl and Python based on SWIG with some examples explained in detail, and share the experience of developing native implementations of BioC functionality in Python, Ruby and the Go language.
BioC Application Architecture
To reach high interoperability and reusability in NLP and text processing tasks, BioC is designed as a simple workflow based on an XML format ( 1 ). The BioC workflow uses two connector modules for XML input/output, as shown in Figure 1 . In a typical BioC application, an ‘Input Connector’ is created to gain access to XML input from a file or network stream. This input is converted to data encapsulated in BioC data classes through an XML parser. The BioC data classes then provide various methods to retrieve data for the data processing stage, which allows the functionality of an application. Modify methods of BioC data classes can be used to update BioC data objects, or new data objects can be created. Finally, the output connector writes the new or updated data in BioC format.
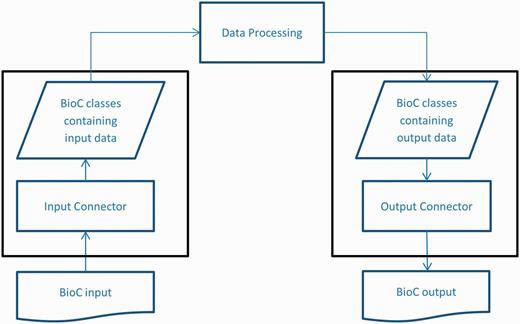
BioC workflow diagram.
This design separates the data processing stage and the Input/Output connectors, which enables the data processing stage to focus on processing biomedical data, without concern for the format of the Input/Output files. The flexible architecture also allows input stage, data processing stage and output stage to be decoupled as needed.
SWIG: a BioC C++ Wrapper for Perl and Python
A BioC C++ SWIG interface
SWIG is a software development tool that we use to connect the core BioC library written in C++ with a variety of high-level programming languages in which BioC users may develop various applications. SWIG is able to support >20 programming languages (Lisp, Octave, R, C#, Lua, etc.), including Python and Perl. Our experience with SWIG indicates that SWIG works well with the BioC core C++ implementation by providing quick development and deployment for BioC users with the supported languages.
SWIG streamlines the process of building a target language module from the C++ code base, as shown in Figure 2 . Specifically, if we take Python as example target language, the BioC C++ header files ( BioC.hpp, BioC_libxml.hpp, BioC_util.hpp ) are processed by SWIG, which extracts the declarations in these header files to create wrapper codes in both the target language Python ( BioC_full.py ) and C++ ( BioC_full_wrap.cpp ), as guided by BioC_full.i , an interface file created manually. The wrapper code in the target language defines proxy classes for the underlying C++ classes, as well as a variety of customizations to suit the specific target language features. The original BioC source code files ( BioC.cpp, BioC_libxml.cpp, BioC_util.cpp ) are also compiled by a C++ compiler to generate object files, in the same way as building a pure C++ application. These BioC object files are linked with the object file ( BioC_full_wrap.o ) compiled from C++ wrapper code to create the BioC module for the target language ( BioC_full.so ).
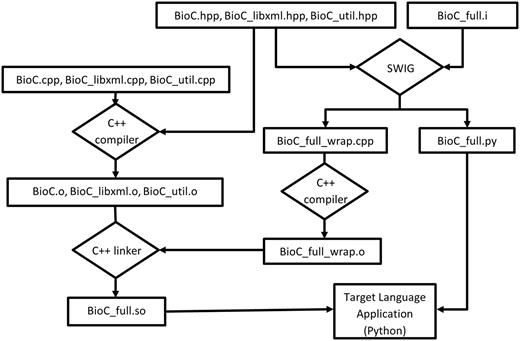
Building BioC modules with SWIG (for Python).
In the target language programming environment, the wrapper code ( BioC_full.py ) provides the interface to BioC functionalities in the target language, while the BioC module ( BioC_full.so ) performs these functionalities. To facilitate seamless data flow across different programming languages, proxy class objects wrapping C++ BioC classes are initialized in the target language, and data are read from BioC files and deposited into C++ BioC class data structures. The application in the target language obtains access to the same data stored in the C++ BioC data structures via methods of BioC classes defined in the wrapper code ( BioC_full.py ) to process the data, as shown in Figure 3 . Because the BioC core C++ implementation is based on libxml2 ( http://xmlsoft.org/ ), all SWIG extension modules share the same XML interface with BioC files. BioC users can build SWIG extensions for their desired target language on their systems equipped with libxml2 (please refer to Readme.txt in BioC SWIG package).
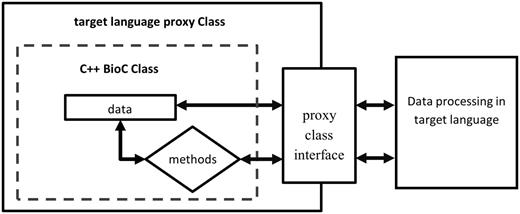
Access C++ BioC class through target language proxy class wrapper interface.
Using SWIG BioC in Perl and Python
To illustrate the key points of using a BioC module in target languages, we use Perl code as an example ( Figure 4 ). Based on BioC C++ version 1.0 ( http://source forge.net/projects/bioc/files/BioC_C%2B%2B_1.0.tar.gz/download ), SWIG version 2.0.4 ( http://sourceforge.net/projects/swig/files/swig/swig-2.0.4/ ) produces BioC_Perl.so and BioC_Perl.pm . BioC_Perl.pm contains the Perl proxy classes for the BioC C++ classes, and BioC_Perl.so provides the executable implementation of BioC classes.

Perl code accessing BioC data (tested with Perl 5.8.8).
After importing the Perl BioC module, we are ready to initialize BioC wrapper objects. The Connector_libxml class object ($xml) enables opening an XML file and reading the contained data into a Collection class object ($collection) . Because libxml functionalities are encapsulated inside the BioC module, the Perl application can focus on processing BioC data. Then we can iterate through the BioC file by visiting each document ($dcm) within the collection, each passage ($psg) within each document and each annotation ($ann) and relation ($rel) within each passage. While iterating through the XML file, the data members defined in BioC classes (e.g. { id } of Document class) can also be accessed directly or through methods via Perl wrapper objects. In addition to reading the whole XML file into ($collection) before processing, BioC also provides a read_next(document) method for one-document-at-a-time access.
In addition to the data read into the BioC class data structure from BioC files, new data can be added to the BioC classes. One such example is the { infons } data structure in a number of BioC classes, which is a C++ std::map container template mapping a key string to a value string. The mapped value string can be retrieved via the get() method of the { infons } data structure with ELEMENT KEY string [and updated via the set() method].
After data are extracted from XML format and then processed by Perl code, the original collection class object can be modified in a fashion similar to reading BioC data classes. The updated collection class object can be saved in XML format, ready to be accessed by another BioC application. The Perl BioC module has been successfully used by the NatLAb abbreviation system ( 6 ) to make it BioC compatible ( 7 ).
Figure 5 shows Python code executing the same task as Perl code. Compared with Perl, BioC data classes interface with native Python structures more naturally, which improves convenience and ease of development. Unlike in Perl, the C++ std::map container is treated as a Python dict object, and a mapped string can be accessed as dict[key]. The example code demonstrates several ways to loop over BioC data. Users should note that some BioC class methods may work differently from corresponding Python methods. For example, the get() method of the dict object should be protected by a preceding key in dict expression in the BioC Python module, whereas the get() method in the Python dict object can return the default value for a missing key.

Python code accessing BioC data (tested with Python 2.5.1).
Normally BioC core C++ library users read and update BioC data using a reference, which operates directly on the BioC class data structure. BioC modules in Python and Perl, in addition to operating on BioC data directly with a reference, also support read-only access to BioC data by implicitly creating a separate copy of BioC data, as shown in Figure 5 . BioC module users can choose the appropriate access mechanism based on the nature of their applications. For example, in an application that modifies BioC data, a better practice may be to create new data objects with updated or new data when memory capacity permits.
Python: a Native Python Implementation of BioC (PyBioC)
For some BioC users, a native BioC library is preferred when complete compatibility is required in the application. The ability to extend native BioC classes within one language context also allows BioC users to add more functionality to original BioC classes through inheritance. The Ontogene team developed the PyBioC library to recreate the functionality of the already available libraries in C++/Java. However, Python conventions are adhered to where suitable. For example, getter or setter methods are avoided for internal variables of the classes provided in PyBioC. Basically the library consists of a set of classes representing the minimalistic data model proposed by the BioC community. Two specific classes (BioCReader and BioCWriter) are available to read in data provided in valid BioC format and to write from PyBioC objects to valid BioC format. Validity is ensured by following the BioC DTD publicly available.
The library is available on a public github repository ( https://github.com/2 mh/PyBioC), where example programs can be found. These programs read in and write to BioC format and can tokenize and stem a given BioC input file using the Natural Language Toolkit (NLTK) library ( http://nltk.org/ ). As an example application, we also provide the integration of a standard word stemmer in PyBioC ( https://github.com/2mh/PyBioC/blob/master/src/stemmer.py ). These programs were tested using Python 2.7.6. Figure 6 shows Python code using PyBioC to execute the same task as in the SWIG BioC section.

PyBioC code accessing BioC data.
PyBioC enables the biomedical text mining community to work with BioC documents using a native implementation of the BioC library in the Python programming language. PyBioC is available as open-source under the simplified BSD license. We welcome further contributions and additions to this work. This Python implementation of the BioC core is available at https://github.com/2mh/PyBioC/tree/master/src/bioc.
Go: A Go Implementation of BioC (go_bioc)
Go is a new language from Google developed by Ken Thompson and Rob Pike, who are known for UNIX, C and Plan 9. Its features include the convenience of type inference and goroutines for concurrency. As their Web page ( http://golang.org/doc/ ) says, ‘It's a fast, statically typed, compiled language that feels like a dynamically typed, interpreted language’.
In Go, XML data can be marshaled and unmarshaled simply by using struct tags. One limitation is that there is no direct way to exchange information between XML and a map, as used to implement infons in other languages. So Go BioC objects hold both an Infons map and an InfonStructs slice. The XML data are marshaled and unmarshaled out of and in to the InfonStructs slice. The data are moved into or out of the Infons map after reading or before writing. This is a small inefficiency because the amount of infon data are small. Reading or writing a BioC collection document at a time does require lower-level interaction with the XML parser for the collection itself. The individual documents can still be written and read with the direct marshal and unmarshal functions. So far we are pleased with the language and expect to continue our experiments.
An example of Go code executing the same task as in the SWIG BioC section is shown in Figure 7 . An important thing to notice about the Go sample code is the option to iterate over the data or an index to the data. Although iterating over the data can lead to slightly simpler code, it implies a copy of the data. Especially in the presence of parallel processing, this might be an advantage. However, in many cases, accessing the data via an index will be more efficient.

Go code accessing BioC data (tested with Go 1.1.2).
Ruby: A Ruby implementation of BioC (simple_bioc)
Ruby is a general-purpose programming language ( http://www.ruby-lang.org ), which has been developed with a focus on simplicity and productivity, similar to Perl and Python. Ruby has received attention in recent years because of Ruby on Rails ( http://rubyonrails.org/ ) for the rapid development process for modern Web sites. Ruby is also useful for fast prototyping or verifying new ideas because it provides a simple approach to software development, and there are many free libraries available in various domains. As mentioned above, BioRuby ( 4 ) and BioGems ( 5 ) are useful libraries for bioinformatics and biomedical applications.
As a Ruby BioC library, the main goal of the simple_ bioc library is to provide a simple and easy way to parse and build the BioC format in Ruby. The simple_bioc library is publicly available from http://rubygems.org/gems/simple_bioc and it can be installed by one command: gem install simple_bioc. Parsing and/or building a BioC document can also be performed by a single line of program code. The source code is available from https://github.com/dongseop/simple_bioc under the MIT license, which allows BioC users to modify or embed simple_bioc in their Ruby applications. The documentation ( http://rubydoc.info/gems/simple_bioc/0.0.3/frames ) includes a usage guide, sample code and a full API reference.
The main modules of simple_bioc were originally implemented for handling BioC documents in BioQRator ( 8 ), a web-based interactive biomedical literature curating system. Simple_bioc has been factored out so that it can be released as an open-source contribution to the BioC and Ruby communities. The reader new to Ruby will note the unique approach to iteration Ruby offers. Although not the traditional approach of many common languages, Ruby offers a number of flexible iteration methods. As with many scripting languages, Ruby values are references, so no unnecessary copying occurs. Figure 8 shows how BioC works in Ruby.

Ruby code accessing BioC data (tested with Ruby 2.0.0).
Conclusions
We describe the extension of BioC beyond C++ and Java programming languages, enabling convenient handling of BioC documents in other languages. SWIG allows the original behavior of C++ BioC classes to be available in scripting language contexts by reusing the same C++ source code. Currently, different languages have different features and capabilities, which are exposed to varying extents by SWIG ( http://www.swig.org/Doc2.0/ ). For example, Python is demonstrated to offer a much richer interaction with the C++ data structures than is available in Perl. However, we believe the current BioC extensions built with SWIG will satisfy most demands of the biomedical text mining community.
A native BioC implementation provides more natural interaction and integration with the language. Native implementations of BioC in Go, Python and Ruby are presented. As expected these fit in well with other language features and behave more as expected by developers in each language. This article demonstrates that BioC can be realistically extended to any language, which is supported by SWIG or an XML parser. These BioC implementations are freely available through the BioC Web site ( http://bioc.sourceforge.net ).
Funding
The research at NCBI was supported by the Intramural Research Program of the National Institutes of Health, National Library of Medicine. The OntoGene group at the University of Zurich is partially supported by the Swiss National Science Foundation (grants 100014-118396/1 and 105315-130558/1). D.K. was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2012R1A1A2044389 and 2011-0022437).
Conflict of interest . None declared.
References
Author notes
Citation details: Liu,W., Islamaj Doğan,R., Kwon,D., et al. BioC implementations in Go, Perl, Python and Ruby. Database (2014) Vol. 2014: article ID bau059; doi:10.1093/database/bau059

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}