





Abstract
Malignant triton tumors (MTTs) are a rare subtype of malignant peripheral nerve sheath tumors characterized by rhabdomyoblastic differentiation, often associated with neurofibromatosis type 1 (NF-1). Primary MTTs of the bladder are exceptionally uncommon, with few documented cases. We report a case of a 75-year-old male without neurofibromatosis type 1 history presenting with hematuria and urinary obstruction. Imaging revealed a 6.5 × 4 × 2 cm invasive bladder mass, and histopathology confirmed a high-grade spindle cell neoplasm with biphasic neurogenic (S100-positive) and rhabdomyoblastic (desmin- and myogenin-positive) differentiation, consistent with a diagnosis of MTT. Despite radical cystoprostatectomy, the patient succumbed to postoperative complications. This case highlights the aggressive behavior, diagnostic challenges, and poor prognosis of bladder MTTs, emphasizing the critical role of immunohistochemistry in distinguishing them from other spindle cell neoplasms. The rarity of this presentation underscores the need for further research to elucidate its pathogenesis and optimize therapeutic strategies.
Introduction
Malignant triton tumors (MTTs), a rare subtype of malignant peripheral nerve sheath tumors (MPNSTs), feature rhabdomyoblastic differentiation. These tumors form 5% to 10% of soft tissue sarcomas and are often linked to neurofibromatosis type 1 (NF-1), with up to 50% of cases in NF-1 patients [1]. MTTs are highly aggressive, with frequent local recurrence and poor prognosis [2]. Primary bladder MTTs are extremely rare, with few reported cases. This report describes a primary bladder MTT, focusing on its histopathological features and aggressive nature.
Case report
A 75-year-old male with no known history of NF-1 presented with hematuria and urinary obstruction for 3 months. Contrast-enhanced CT imaging revealed a heterogeneous mass measuring 6.5 × 4 × 2 cm with irregular borders and central necrosis, extending into the perivesical fat and abutting the prostate (Fig. 1).
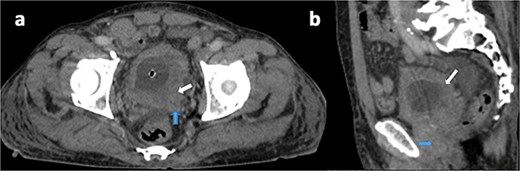
Axial (a) and sagittal (b) contrast-enhanced CT views in portal phase showing a diffuse and irregular thickening of bladder wall, pronounced on the left posterolateral side (white arrow), with invasion of the seminal vesicles and the prostate (blue arrow).
Cystoscopy and biopsy were performed, and histopathological examination revealed a high-grade spindle cell neoplasm with features consistent with MPNST.
Due to local invasion, a radical cystoprostatectomy was performed. However, the patient developed postoperative complications, including sepsis and multiorgan failure, and succumbed to these complications 10 days post-surgery.
On gross examination, the tumor measured ~6.5 × 4 × 2 cm, pearly white, fibrotic, and firm with focal yellow discoloration (Fig. 2).

Gross appearance of the bladder tumor after sectioning, showing a solid whitish lesion invading the bladder wall.
Microscopically, the lesion exhibited a biphasic pattern characterized by a spindle cell component with fascicular growth and interspersed rhabdomyoblastic elements. The spindle cells were elongated with hyperchromatic, wavy nuclei, and scant cytoplasm, arranged in a storiform or herringbone pattern.
The rhabdomyoblastic component consisted of larger, polygonal cells with abundant eosinophilic cytoplasm and eccentric nuclei. Mitotic activity was brisk, with up to 10 mitoses per 10 high-power fields, and focal areas of tumor necrosis were noted. No epithelial or glandular structures were identified within the lesion.
Immunohistochemically, the spindle cell component showed focal positivity for S100 protein (PS100), with staining limited to scattered clusters of cells, highlighting the neural crest-derived Schwannian elements. The rhabdomyoblastic component showed focal positivity for desmin and myogenin, with strong cytoplasmic staining for desmin and nuclear staining for myogenin in the larger polygonal cells, confirming skeletal muscle differentiation (Fig. 3).
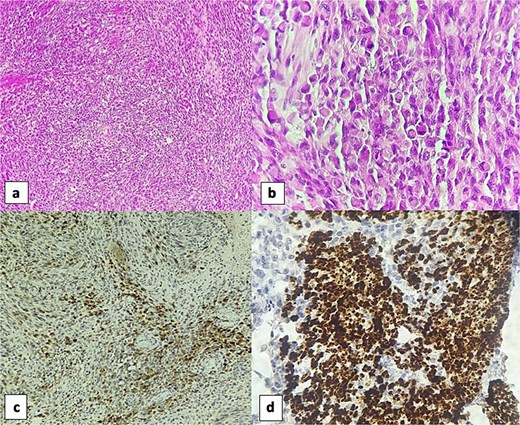
Sections of the vesical tumor revealed a proliferation of spindle cells arranged in fascicles (H&E, 4×) (a). The rhabdomyosarcomatous component was evident with numerous rhabdomyoblasts (H&E, 40×) (b). Immunohistochemically, the spindle cell component showed focal positivity for S100 protein (10×) (c). Rhabdomyoblastic component was positive for desmin (40×) (d).
Additional markers, including Melan-A and HMB-45, were uniformly negative, excluding melanocytic differentiation, while cytokeratins (AE1/AE3, CK7) were also negative throughout, ruling out epithelial or carcinoma-like origins.
Discussion
MTTs, a rare subtype of MPNSTs, combine neurogenic elements from Schwann cells and rhabdomyoblastic components with skeletal muscle differentiation. Typically arising along peripheral nerves in the extremities, trunk, or head and neck, primary bladder MTTs are extremely rare [3]. This rarity creates significant diagnostic and therapeutic challenges, as seen in our patient without NF-1 history. MTT pathogenesis is poorly understood, but 50%–70% of cases are linked to NF-1, indicating a genetic predisposition due to NF-1 tumor suppressor loss [2].
In NF-1-associated MTTs, rhabdomyoblastic differentiation is thought to reflect aberrant mesenchymal differentiation within a neurogenic tumor. Yet, in sporadic cases like ours, the absence of NF-1 raises questions about alternative oncogenic drivers. Molecular testing for NF-1 mutations was not performed, but such analysis could clarify sporadic MTT pathogenesis. Environmental factors, somatic mutations, or prior local insults (e.g. chronic inflammation or irradiation) may contribute, though our patient had no such history. The bladder’s mesenchymal-rich environment may predispose to such divergent differentiation, but this remains unproven due to the paucity of reported cases. Diagnosing primary bladder MTT is challenging due to its histological overlap with other spindle cell neoplasms, including leiomyosarcoma, rhabdomyosarcoma, sarcomatoid urothelial carcinoma, or inflammatory myofibroblastic tumor. In our case, a high-grade spindle cell tumor with local invasion prompted a broad differential diagnosis. Definitive diagnosis relied on histopathological examination revealing dual neurogenic (S100-positive) and rhabdomyoblastic (desmin- and myogenin-positive) features, a hallmark of MTTs. This underscores the essential role of immunohistochemistry in distinguishing these tumors from mimics, particularly in atypical locations like the bladder where clinical suspicion may be low. Imaging findings, such as the heterogeneous mass with irregular borders and perivesical extension observed in our patient, are nonspecific and further emphasize the need for tissue sampling [4].
The prognosis of MTTs is generally poor, with aggressive local behavior and a high propensity for metastasis, often to the lungs or bones [3]. Bladder involvement may exacerbate this outlook due to the organ’s anatomical constraints and proximity to critical structures, complicating complete surgical resection. In our patient, the tumor’s invasion into adjacent tissues necessitated radical cystoprostatectomy, with adjuvant radiotherapy planned to address residual disease risk, but not administered due to postoperative complications. However, the efficacy of such multimodal approaches in MTTs remains anecdotal, as most data derive from extracranial or extremity-based cases. Chemotherapy, including anthracycline-based regimens, has shown limited success in advanced MTTs, and targeted therapies remain investigational, hindered by the tumor’s molecular heterogeneity.
The rarity of bladder MTTs limits comparative analysis, with very few cases reported in the literature. The following table summarizes the clinical and histopathological features of our case (Table 1) [5].
Features of primary MTT case of the bladder
| Case | Age | Sex | Localization | NF-1 status | Treatment | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| 1 | 2 months | F | Primary bladder | Yes | Local excision | Recurrence, died of brain metastasis | Daimaru et al. [5] |
| 2 | 75 years | M | Primary bladder | No | Radical cystoprostatectomy | Death at 10 days | Current case |
The rarity of bladder MTTs prevents standardized treatment guidelines, requiring extrapolation from MPNST protocols or individualized approaches. Our case underscores the need to document such rare cases to clarify the clinical spectrum of MTTs and refine diagnostic criteria. Future genomic profiling studies may reveal unique molecular signatures in bladder MTTs, potentially enabling personalized therapies.
In conclusion, this case of a primary MTT of the bladder illustrates the diagnostic complexities and treatment challenges posed by this rare malignancy. Its aggressive nature and unusual location underscore the need for heightened awareness among clinicians and pathologists, as well as the urgent call for collaborative research to improve outcomes in this difficult disease entity.
Conflict of interest statement
None declared.
Funding
None declared.
Data availability
Not applicable.

{kind=link}
{kind=link}
{kind=link}