





Abstract
Primary breast lymphoma (PBL) is a rare neoplasm, accounting for ˂1% of all breast cancers. Most cases are of B-cell phenotype and typically present as a painless breast mass. Imaging findings are often non-specific, mimicking benign pathology, and diagnosis is confirmed through histopathological analysis. While no standardized treatment guidelines exist, surgery plays a limited role, with the cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) regimen, with or without radiotherapy, being commonly employed. Here, we present a rare case of PBL in a middle-aged African female involving the left breast, predominantly toward the axillary tail. She presented with a painless mass, and the diagnosis was confirmed by histology. The patient is currently undergoing CHOP chemotherapy with significant regression of clinical symptoms and no adverse effects reported. This case underscores the importance of early referral to a tertiary center for histopathological evaluation and timely initiation of appropriate therapy.
Introduction
Primary breast lymphoma (PBL) is an exceptionally rare malignancy, accounting for 0.04%–0.7% of all breast cancers and ˂2% of extranodal non-Hodgkin lymphomas (NHLs) due to the scarcity of lymphoid tissue in the breast [1]. PBL is defined as lymphoma localized to the breast without systemic involvement, with most cases presenting a B-cell phenotype. Typically manifesting as a palpable mass, its imaging features are nonspecific and may mimic benign lesions [2]. There is no consensus on the optimal management approach, which may include surgery, radiotherapy, chemotherapy, and immunotherapy [3]. Here, we present a case of a 42-year-old woman diagnosed with PBL in the axillary tail of the breast.
Case presentation
A 42-year-old female presented with a progressively enlarging left axillary mass over four months. She reported no breast pain, nipple discharge, or skin changes. Her medical history included combined oral contraceptive use, with no family history of breast or gastrointestinal cancers.
On examination, her ECOG performance status was 1. She was mildly pale, with a 13 × 11 cm hard, fixed mass in the upper outer quadrant of the left breast extending into the axillary tail. Ipsilateral supraclavicular lymphadenopathy was noted, while the contralateral breast was unremarkable.
A computed tomography (CT) scan revealed a necrotic soft tissue mass (13.5 × 9.8 × 8.2 cm) infiltrating anterior chest wall muscles, subscapularis muscle, adjacent skin, and subcutaneous fat, with ipsilateral axillary and supraclavicular lymphadenopathy, consistent with T4N3M1 breast cancer (Fig. 1). Laboratory tests showed microcytic hypochromic anemia (hemoglobin: 9.3 g/dL) and a normal leukocyte and platelet count. A biopsy of the mass with immunohistochemistry confirmed diffuse large B-cell lymphoma (DLBCL) with strong CD20 positivity (Fig. 2).
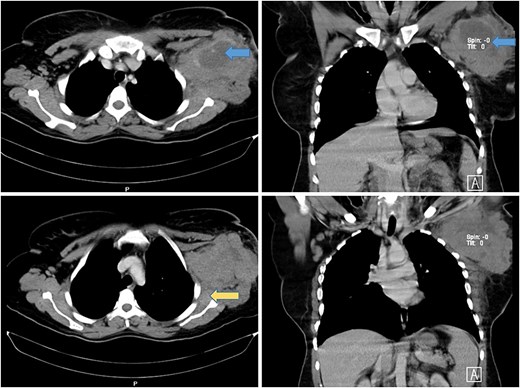
Axial and coronal CT scan of the chest showing a large mass in the left axillary tail of the breast with areas of necrosis (blue arrows) with involvement of anterior chest wall muscles and infiltration of the left subscapularis muscle (yellow arrow).
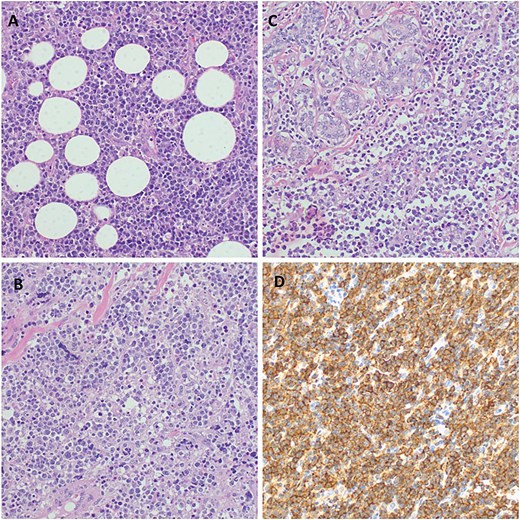
Histopathology of the breast tumor displaying diffuse aggressive high-grade lymphoma characterized by a dense malignant lymphoid infiltrate of fibro-adipose tissue, H&E staining at 40× original magnification (A). Photomicroscopy of the tumor highlighting a population of diffuse large lymphocytic cells that are moderately pleomorphic with scant dense cytoplasm and have enlarged hyperchromatic nuclei, with irregular contours, coarse chromatin, and prominent nucleoli invading the fibrous and skeletal muscles; H&E staining at 100× original magnification (B). Infiltration of the breast tissue by a diffuse large cell lymphoma and surrounding breast parenchyma obliterating mammary gland structures, H&E staining at 100× original magnification (C). Photomicroscopy showing a strong membrane immune-expression of the tumor cells with CD 20; IHC staining a 100× original magnification (D).
After multidisciplinary team (MDT) discussion, the patient was initiated on the R-CHOP regimen (cyclophosphamide, doxorubicin, vincristine, prednisone). She is under follow-up in the oncology clinic, where the mass has significantly reduced in size.
Discussion
First described in 1959, PBL is a rare neoplasm due to the limited lymphoid tissue in the breast [4, 5]. Its incidence is estimated at 1.35 per 100 000 cases from 1975 to 2017 [5]. It predominantly affects women, with an average age of onset ⁓60 years, though it can range from 9 to 85 years. Male cases are exceedingly rare [6, 7].
Clinically, PBL usually presents as a painless, rapidly enlarging, unilateral breast mass, though 1%–14% of cases show bilateral involvement. Skin and nipple changes are uncommon. PBL is more aggressive during pregnancy and lactation, often presenting bilaterally and advancing rapidly, likely due to hormonal influences [6, 8].
Two hypotheses exist regarding PBL’s origin: lymphoma arising from intramammary lymph nodes (widely accepted) and connective tissue origin involving undifferentiated mesenchymal cells. Most PBL cases are NHLs, predominantly high-grade B-cell lymphomas like DLBCL (45%–79%), as seen in our patient. Other less common types include follicular lymphoma, mucosa-associated lymphoid tissue lymphoma, and Burkitt lymphoma. Hodgkin lymphoma involving the breast is extremely rare [9, 10].
The diagnostic criteria for PBL were first defined in 1972 by Wiseman and Liao and refined by the International Extranodal Lymphoma Study Group. The latter requires:
No primary tumor outside the breast.
No history of prior lymphoma or systemic involvement at diagnosis.
Pathologic evidence of lymphoma in breast tissue.
Ipsilateral lymph node involvement occurring simultaneously with the primary tumor [5, 8, 9].
Imaging features of PBL are nonspecific and may resemble adenocarcinoma. Mammography typically shows a solitary, hyperdense mass with circumscribed or indistinct margins and absent calcifications. MRI may reveal round or oval masses with variable enhancement patterns [9]. In low-resource settings like Tanzania, CT scans remain a valuable diagnostic tool despite the superiority of magnetic resonance imaging (MRI) and positron emission tomography/computed tomography (PET/CT).
Histologically, PBL tumors are categorized into large-cell B-cell lymphomas, monocytoid B-cell lymphomas, and undifferentiated lymphomas. Immunohistochemical markers such as CD20 and Ki-67 aid in diagnosis and grading. Most breast lymphomas are of B-cell origin, with DLBCL accounting for 40%–70% of cases [9].
Management strategies for PBL range from surgery to combination chemotherapy and radiotherapy. However, there is no established standard of care. Mastectomy is generally not supported, as it neither improves survival nor reduces recurrence risk [6]. Similarly, axillary dissection offers no therapeutic advantage. Chemotherapy, particularly the R-CHOP regimen, combined with radiotherapy, has demonstrated the most favorable outcomes [8].
Prognosis depends on lymphoma type, grade, and stage. PBL generally has a better prognosis than secondary breast lymphoma due to its localized nature. Low-grade PBL has higher survival rates and better local control, whereas high-grade PBL is more aggressive with poorer outcomes [9].
This case highlights the rare occurrence of PBL in our region, representing only the second reported case in Tanzania. The introduction of MDT discussions and immunohistochemistry has advanced breast cancer diagnostics locally, enabling better management of cases like PBL. This report underscores the need for further research into PBL management and outcomes to align with global standards.
Acknowledgements
The authors would like to thank the patient for permission to share her medical information to be used for educational purposes and publication.
Author contributions
Patient care and methodology: TEM, Conceptualization and writing original manuscript: MS, Data analysis: AM, Data curation: AM and DM, Editing, writing and supervision: JL. All authors have read and approved the final version of the manuscript.
Conflict of interest statement
None declared.
Funding
No funding or grant support.
Informed consent
Written informed consent was obtained from the patient for publication for this case report; additionally, accompanying images have been censored to ensure that the patient cannot be identified. A copy of the consent is available on record.
References
Author notes
Theresia E. Mwakyembe and Mujaheed Suleman contributed equally to this article.

{kind=link}
{kind=link}