





Abstract
Primary adrenal angiosarcoma is an aggressive and extremely rare malignancy. Given this, there is a paucity of reported cases and clinical guidelines for its management. Here, we present a case report of a 70-year-old male diagnosed with primary adrenal angiosarcoma. He underwent surgical resection with curative intent and is currently undergoing surveillance with no evidence of local recurrence or metastases after 7 months. This case report contributes to the literature by increasing our knowledge regarding this rare entity.
Introduction
Angiosarcomas account for 1% of soft tissue sarcoma presentations [1]. Primary adrenal angiosarcomas are even more infrequent with ~50 cases reported in English language literature to date [2]. We present a case of a 70-year-old male diagnosed with primary adrenal angiosarcoma. The diagnosis of adrenal angiosarcoma is challenging in part due to its rarity and its radiographic similarities with adrenal cortical carcinoma (ACC). Given the low prevalence of this disease, high-level randomized control trials are unfeasible. Consequently, case report publications are needed to inform our knowledge and aid in clinical diagnosis and management.
Case report
A 70-year-old male presented with a forty-pound weight loss and left flank pain in the preceding 6 months. Past medical history was significant for coronary artery disease, hypertension, well controlled non-insulin dependent diabetes, anemia, colitis, and increased cholesterol. He denied any previous abdominal surgeries. Current medications included ferrous gluconate, rosuvastatin, bisoprolol, mesalamine, gliclazide, metformin, and vitamin D. He is an ex-smoker and denies current alcohol use.
On physical exam his vitals were stable and within normal limits. In the left upper quadrant, he was tender and had a vaguely palpable mass.
Laboratory investigations revealed his hemoglobin was chronically low at 83 g/L and his white blood cell count (WBC) was elevated at 19 × 109/L. He had a WBC elevated at ~20 × 109/L during the preceding 6 months. His serum potassium, 24-h urine metanephrine, 24-h urine cortisol, plasma metanephrine, plasma normetanephrine, and aldosterone to renin ratio were within normal limits. The dehydroepiandrosterone sulfate was slightly low. Interestingly, a 1 mg dexamethasone suppression test overnight yielded an 8 am cortisol level of 98.5 nmol/L, indicating that cortisol was not fully suppressed with 1 mg of dexamethasone.
Computed tomography (CT) abdomen/pelvis demonstrated a 20.5 × 13.7 × 13.9 cm hypoenhancing lobulated mass in the left upper quadrant with stippled areas of calcification within the mass (Fig. 1). There was a maintained fat plane between the mass and kidney. The left adrenal gland was not definitely visualized. Given the radiologic findings, the presumptive diagnosis of an ACC was made. There was no evidence of metastatic disease or local invasion into the renal vein or vena cava. A normally enhancing right kidney was seen.
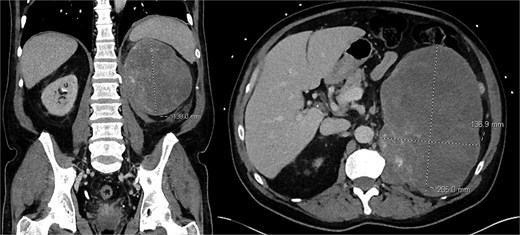
Pre-operative CT scan of abdomen/pelvis findings of a left upper quadrant abdominal mass.
The case was discussed at multidisciplinary soft tissue rounds and upfront surgery was recommended. Based on the working diagnosis of a non-functional ACC, percutaneous biopsy was not obtained as it would not affect management. Even if this tumor was a high-grade sarcoma neoadjuvant radiotherapy would not have been recommended based on the STRASS trial [3].
A midline laparotomy was performed, revealing no evidence of liver metastasis or carcinomatosis. The lesion was found to involve the capsule of the left kidney and resection of the left retroperitoneal mass and left radical nephrectomy was performed without complication.
Final pathology revealed an angiosarcoma of the adrenal gland. Sections showed a predominantly hemorrhagic and necrotic mass with residual vascular forming channels within the adrenal gland (Fig. 2). Cells were pleomorphic spindled to epithelioid and atypical (Figs 3 and 4), with a mitotic count of 12/10 high-power fields. Immunohistochemistry showed positivity for ERG (Fig. 5), CD34, and focal pankeratin and negativity for SF-1, SOX10, Cathepsin-K, and chromogranin. There was no lymphatic or vascular invasion identified, and the margins were uninvolved by the tumor. The left kidney was negative for tumor, and the background adrenocortical parenchyma was benign.
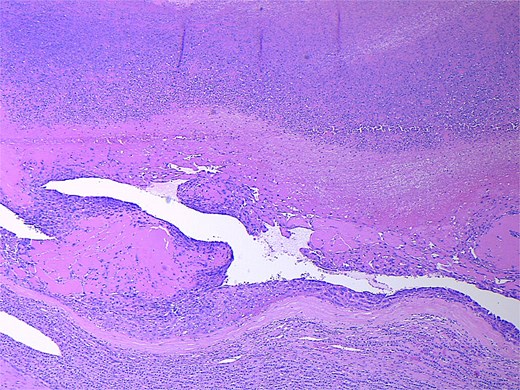
Low power magnification (2×) Hematoxylin and eosin (H&E) showing the angiosarcoma within residual non-neoplastic adrenal cortical tissue. Necrosis is also present.
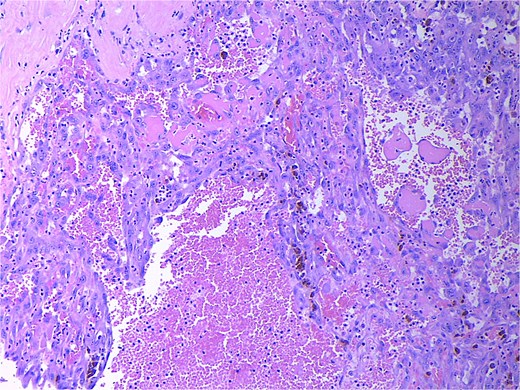
High power magnification (20×) H&E showing malignant cells forming vascular channels with atypia and multiple mitotic figures.
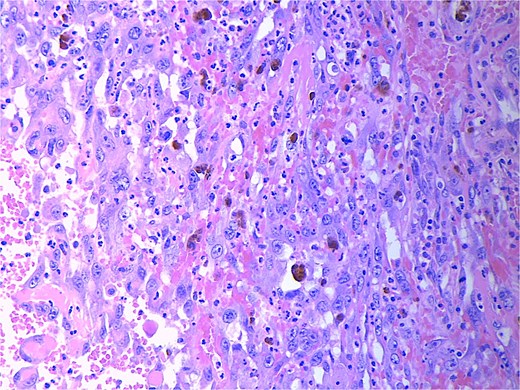
High power magnification (40×) H&E showing malignant cells forming vascular channels with atypia.
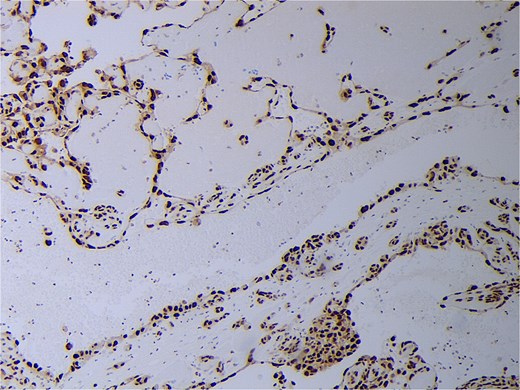
Angiosarcoma staining positive with ERG immunohistochemistry.
His post-operative course was uncomplicated, post-operative creatinine was normal, and he was discharged on post-operative day six. His most recent CT chest, abdomen, and pelvis performed 7 months post-resection showed no evidence of residual, or recurrent local or metastatic disease. Based on the recommendations of medical oncology he is undergoing surveillance with CT chest and CT or magnetic resonance imaging (MRI) abdomen/pelvis every 3–6 months in the initial 5-year post resection time frame. Given the negative margins there is no current plan for further systemic therapy unless there is evidence of recurrence.
Discussion
Primary adrenal angiosarcoma is an aggressive malignancy with a median survival of 1.5 years [4]. While there are no pathognomonic radiological findings, CT imaging is beneficial in the investigation and management. In this case, CT imaging demonstrated a hypoenhancing lesion with calcifications. Adrenal angiosarcomas and ACC have radiographic similarities such as the inclusion of calcifications [5]. While a MRI was not performed in this case, some MRI features may include foci of necrosis and heterogenous enhancement [5]. Locally advanced ACC often presents with local invasion/tumor emboli to the renal vein or vena cava as seen in 25% of cases [6]. At time of diagnosis, 70% of patients with ACC have locally advanced or metastatic disease (Stage 3–4 disease) [7].
In this case, pleomorphic spindled to epithelioid cell morphology was found. In the literature, epithelioid cells were discovered most frequently with the notable exception of a small number of published cases that reported spindle cell morphology [4]. On immunohistochemistry, markers for ETS-related gene (ERG), focal pankeratin, and CD34 were positive in our case, which is consistent with the literature and further supports the diagnosis [2, 4, 8]. In addition to ACC, other differentials include metastatic carcinoma, sarcomatoid melanomas, epithelioid hemangioendothelioma, and anastomosing hemangioma [2, 9]. Therefore, morphologic findings of atypia, necrosis, and increased mitotic counts, immunohistochemical workup, and clinicoradiologic correlation are required. In this case, there were no other sites of disease, leading to the conclusion of a primary adrenal angiosarcoma.
There are no clinical practice guidelines for primary adrenal angiosarcoma. Surgery remains the only option for curative intent. Unfortunately, the natural history of this devastating disease consists of a high local and distal recurrence rate with a 30% 5-year survival rate [4]. Palliative chemotherapy is often utilized for metastatic disease and adjuvant chemotherapy and radiation have been utilized in a variety of protocols throughout the literature [4, 5, 10].
Conclusion
Primary adrenal angiosarcomas are rare thereby making diagnosis and management challenging. Surgery remains the gold standard treatment for curative intent for adrenal angiosarcomas. Publication of case reports augments our knowledge of the disease and increasing the quantity of published case reports will aid in the eventual creation of higher-level evidence guiding the diagnosis and management of adrenal angiosarcoma.
Conflict of interest statement
None declared.
Funding
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}