





Abstract
Adenomatoid tumor (AT) is a benign growth that originates from mesothelial cells. Typically, it encompasses the uterus, fallopian tubes, and paratesticular area. One-third of all AT is in paratesticular area, and it accounts for 60% of all benign paratesticular tumors. ATs have been seldom reported as a case report in adrenal gland, liver, pleura, and mediastinal cavity. To date, English literature has only documented 46 instances of adrenal AT. We present the occurrence of a new clinically unexpected case of adrenal AT in a 37-year-old male patient. Computed tomography (CT scan) incidentally revealed the presence of a tumor. To the best of our knowledge, this is the first reported instance of primary adrenal gland AT reported in Saudi Arabia and the 47th instance worldwide in the English-based published literature. It is crucial to thoroughly investigate these tumors, utilizing techniques such as histopathological analysis and immunohistochemical staining to correctly diagnose AT.
Introduction
Almost a century ago, adenomatoid tumors (ATs) were described, but their true nature and pathogenesis have been a topic of discussion for many years. Since they follow a benign pathogenic progression and are frequently incidentally discovered, there is a noticeable scarcity of literature on them. This has resulted in a lack of knowledge regarding their development and their potential connection to other mesothelial pathology. Nevertheless, advancements in their molecular biology have been observed recently. Golden and Ash reported the initial collection of 15 tumors found in the epididymis, testicular tunics, and the serosal surface of the uterus in 1945 [1]. The term ‘adenomatoid tumor’ was initially suggested by the authors. It was their belief that the tumor’s primary component was epithelial and that it tended to create gland-like structures, although its origin remained unclear. The term quickly became widely recognized and remains in use today. Masson and colleagues [2] were the first to accurately suggest that AT originated from the mesothelium, likely stemming from the pelvic serous lining. However, this theory did not gain significant support, mainly due to the absence of reports on extragenital tumors at that time. Craig and Hart [3] likely reported the first tumor outside of the reproductive system in 1969, which was found in the small intestine. With the advent of electronic microscopy in pathology, the mesothelial nature gained increasing support and revealed that cases previously diagnosed solely on morphological characteristics differed upon further examination, as vascular tumors were frequently misidentified as ATs at that time. Most cases reported were related to female genital tract, with particular emphasis on the uterus and fallopian tubes, while the male genital tract mostly consisted of paratesticular tumors. Cases involving the adrenal gland are infrequent, and occurrences in other peritoneal sites, the liver, pleura, or the mediastinum are even more uncommon. Determining the exact epidemiology linked with AT in the adrenal gland can be a difficult task since the majority of published cases are either solitary or in small case series.
Case presentation
This is a 37-year-old male patient with a history of laparoscopic cholecystectomy 1 year ago, gastric sleeve surgery 2 years ago, and left-sided varicocelectomy 3 years ago. Otherwise, he is healthy and presents no complaints, with stable vital signs. During the radiological work-up for cholecystectomy 1 year ago, a right adrenal mass was incidentally discovered. Multi-axial enhanced computed tomography (CT) scan of the abdomen and pelvis with IV contrast administration utilizing adrenal protocol revealed a small, well-defined oval-shaped complex cystic lesion in the right adrenal gland, measuring 3.5 × 2.3 × 3.2 cm with enhancing thick nodular septations measures. The left adrenal gland is unremarkable, with no other lesions identified. Despite recommendations for surgical intervention, the patient opted for conservative management and underwent regular clinic follow-up for 1 year. Subsequently, multisequential and multiplanar abdomen magnetic resonance imaging for adrenal protocol with precontrast and postcontrast administration was performed and showed a stable right multilocular cystic adrenal lesion with high signal intensity in T2-weighted images and intermediate signal intensity in T1-weighted images without restricted diffusion-weighted images (Fig. 1A). The lesion measures 3.6 × 3 × 3.3 cm in Anteroposterior, transverse, and craniocaudal dimensions. Internal septations exhibited subtle enhancement, with no solid component noted. The rest of the abdominal organs’ liver, pancreas, spleen, left adrenal gland, and both kidneys are within normal limits. The patient agreed for right radical adrenalectomy and the operation went well without complications. The removed specimen was received at the pathology department, which was handled with care for opening and overnight formalin fixation. Gross examination and serial section showed a gray-tan well-demarcated nodule, measuring 3.6 × 3 × 2.5 cm, composed of a multi-cystic lesion without necrosis or hemorrhage (Fig. 1B). Microscopic evaluation demonstrated an unencapsulated, demarcated lesion composed of glandular-like spaces ranging from cords of solid cuboidal and low-columnar cells to expanded spaces lined by significantly flattened cells (Fig. 1C). Lymphoid aggregates were seen frequently (Fig. 1D). Areas of spaces separated by delicate septae were seen giving an alveolar appearance (Fig. 1E). There are no mitoses, necrosis, nor atypia present. Clear surgical resection margin identified. Immunocytochemically, tumor cells were strongly positive for Pankeratin, Calretinin (Fig 1F), D2–40, and WT1, while negative for carcinoembryonic antigen (CEA) and vascular markers such as erythroblast transformation specific (ETS)-related gene (ERG), CD34, and CD31. Based on clinical, pathological morphology, and immunohistochemistry, the final diagnosis was consistent with AT of the adrenal gland. The patient remained healthy without complaints during the 10 months of follow-up.
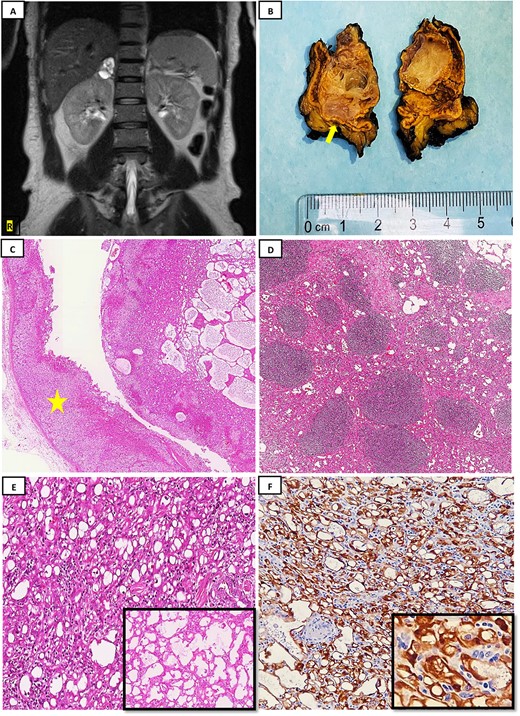
(A) MRI with pre- and postcontrast administration showed stable right multilocular cystic adrenal lesion with high signal intensity in T2-weighted images; (B) gross examination after serial sections reveals a well-demarcated nodule measuring 3.6 × 3 × 2.5 cm, composed of a multi-cystic lesion; a rim of normal adrenal gland is noted to be remaining (yellow arrow); (C): histopathology examination by hematoxylin and eosin stain (H&E) reveals a demarcated lesion composed of micro and macrocytic spaces, with adjacent normal adrenal gland tissue observed (yellow star) (H&E; 4×); (D) lymphoid aggregates are commonly encountered in these tumors as seen here (H&E; 10×); (E) tumor cells composed mostly of microcysts arrangement (H&E; 40×), with the inset showing tumor forming channels giving an alveolar appearance (H&E; 40×); (F) tumor cells show diffuse strong Calretinin positivity (20×), with high power magnification (40×) (inset).
Discussion
The occurrence of ATs in the adrenal gland is rare, with only 46 cases documented in the English literature. Our case represents the first reported instance of primary adrenal gland AT in the Kingdom of Saudi Arabia. Etiology is not well-understood as the adrenal gland does not have a mesothelial lining. There are two theories being proposed for the origin of the tumor. One suggests that mesothelial inclusions may be present as the cell of origin, while the other theory proposes that primitive mesenchymal cells associated with the Müllerian tract may be responsible for the pathogenesis [4]. The majority of adrenal ATs described in published literature are composed of solid tumor growth, with only a small percentage being solid-cystic or fully cystic. Cystic tumor changes raise the possibility of misdiagnosis as lymphangiomas. We believe that the misdiagnosis has been caused by the dilated cystic ‘channels and slit-like’ spaces lined by flattened cells. Although there is a broad age range, ATs commonly appear in individuals in their 40s. Males are more likely to develop tumors, with a ratio of 39:2 compared to females. One potential explanation for the differing roles of mesonephric ducts in male and female embryological development, coupled with the theory that adrenal tumors stem from mesenchymal cells linked to the Mullerian tract, is that these ducts gradually transform into the epididymis duct in males and disappear in females, with the mesonephric ducts receding early on in female development. As a result, it is possible that developing tissue remains present in males, which could account for the prevalence of adrenal tumors in males compared to females [5]. Our study revealed that adrenal ATs are mostly non-functional, asymptomatic, and discovered incidentally during unrelated radiology studies, surgery, or autopsy. Although the symptoms were not specific to the disease, approximately 24% of adrenal AT cases were associated with hypertension. There was only one patient who showed signs of gross hematuria, which was probably caused by the suprarenal mass pressing against the upper area of the kidney [6]. Although adrenal ATs are not associated with immunodeficiency, a case was discovered during autopsy of an individual with acquired immunodeficiency syndrome who had incidentally developed AT. Sadly, this individual passed away due to disseminated coccidioidomycosis infection [7]. Pathologists may face a challenge in diagnosing adrenal AT that may mimic other differentials such as adrenal cortical tumors, lymphangioma, or metastatic adenocarcinoma. The presence of signet-ring–like cells can pose significant challenge as it may indicate the possibility of an adenocarcinoma. Despite the lack of extensive clinical follow-up data, adrenal ATs seem to be benign, much like their genital counterpart. To date, none have shown local disease recurrence nor metastasis [6, 7]. Microscopic evaluation reveals generally demarcated lesion. However, it may extend into various regions such as adrenal capsule, cortex, medulla, or surrounding adipose tissue. The latter is not an indicator of malignancy. AT can exhibit various morphologic architectural patterns such as gland forming or adenoid, tubular, angiomatoid, cystic, solid, and papillary, with most cases manifesting a combination of two or more patterns. Cellular atypia, mitosis, and necrosis are totally absent. Lymphocytic infiltration and aggregates, as in our case, are typically present in most adrenal AT cases [6–8]. Additional histological features identified in the literature include the presence of dystrophic calcification, adipose tissue within the tumor, and ossification [8]. Immunohistochemistry studies demonstrate strong reactivity for cytokeratin AE1/AE3, CAM5.2, CK7, and mesothelial markers such as Calretinin, D2–40 (Podoplanin), WT-1 and HBME-1. GLUT1 may show cytoplasmic reactivity, while vascular markers CD31, CD34, factor VIII are negative. Negative staining is expected for PAX8, CEA, EMA, HMB-45, S-100, smooth muscle actin (SMA) and Desmin. Nearly all cases of adrenal AT show positive expression for D2–40 and Calretinin. We strongly recommend the use of Calretinin, D2–40, and WT-1, as these markers are comparatively specific for identifying ATs. Although ATs in the male and female genital tract are characterized by mutations in the TRAF7 gene, distinguishing them from malignant mesotheliomas, this mutation has not been documented in adrenal ATs in the English literature [9]. Indeed, additional research studies are necessary to investigate the molecular genetics of adrenal ATs.
Conclusion
Adrenal AT is a benign mesothelial neoplasm. Unexpected location such as adrenal gland could be a diagnostic pitfall. AT tend to occur in 40s with noticeable male predilection. Most of these tumors are asymptomatic, incidentally discovered during unrelated radiology, clinical workup, or during autopsy. Multiple growth patterns can be seen with a mixture of more than one in the same tumor, such as glandular, tubular, angiomatoid, micro, and macrocystic solid, and rarely papillary. Adrenal ATs follow an excellent clinical course, and complete surgical excision is the main treatment. No evidence of recurrence and/ or metastasis has been reported to date. Additional research is necessary to elucidate the pathogenesis that links extra-genital locations of AT formation and to establish clear correlations with other associated types of mesothelial lesions.
Conflict of interest statement
None declared.
Funding
None declared.

{kind=link}