





Abstract
Castleman disease (CD) is a rare lymphoproliferative disorder that can be life threatening if left unmanaged in severe cases of inflammatory response. CD should be excluded by thorough workup in cases of lymphadenopathy and splenomegaly of unknown cause. Excisional biopsy of lymph nodes may be required to make a definitive diagnosis. A case of CD manifesting as portal hepatis lymphadenopathy is presented.
INTRODUCTION
Castleman disease (CD) is a rare group of lymphoproliferative disorders causing lymphadenopathy presenting with a wide range of clinical manifestations [1]. The disease was first reported by Benjamin Castleman in the 1950s as mediastinal lymphadenopathy, and further subdivided in the 1960s into variants such as hyaline-vascular, plasma cell (PC) and mixed types [1]. This case report describes a rare case of unknown cause of lymphadenopathy and splenomegaly that was eventually diagnosed as CD by laparoscopic excisional biopsy of an enlarged porta hepatis lymph node.
CASE REPORT
A 28-year-old male presents with a 3-year history of fatigue, night sweats and 10 kg unintended weight loss. He has no other medical conditions and is not on regular medications. He has no family history. Initial examination revealed a palpable liver edge 2 cm below the costal margin and a palpable spleen tip. Preliminary blood results reported microcytic iron deficiency anaemia (Hb 84 g/L), high platelet count (599 × 10^9/L), elevated CRP of 101 mg/L and polyclonal gamma globulin and light chain increase.
A computed tomography (CT) of the chest, abdomen and pelvis was organised and showed splenomegaly of 16.5 cm and suspicion of an enlarged lymph node between the head of the pancreas and the liver (Fig. 1). Magnetic resonance cholangiopancreatography (MRCP) did not further aid diagnosis.
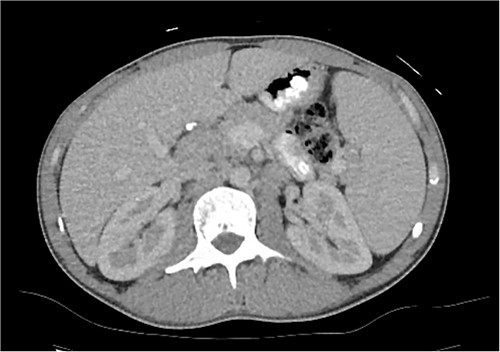
CT axial slice of abdomen and pelvis demonstrating an enlarged lymph node at the porta hepatis and splenomegaly.
Bone marrow biopsy showed PC excess with no definite evidence of clonality favourable for reactive changes. He underwent an endoscopic ultrasound and biopsy, which found an enlarged 27 mm porta hepatis node and several suspicious lymph nodes at the coeliac axis. Fine needle aspiration of the porta hepatis node showed a mixed inflammatory infiltrate with prominent PC. Further blood tests reported IL-6 of 30 units, high IgG and IgA, normal IgM and high IgG1, IgG3 and IgG4 subclasses.
Despite investigations, the diagnosis was still unclear. Because of this, he underwent a laparoscopic excisional biopsy of the porta hepatis mass, situated postero-lateral to the common bile duct and common hepatic duct. This mass was visible on the abdominal CT scan (Fig. 1), yet much less clearly seen on the MRCP, which had been performed more recently. Biopsy results showed marked infiltration of PC with no evidence of Hodgkin like cells or evidence of metastatic malignancy. Human gammaherpesvirus 8 was negative with diffusely positive IgG PC. He was diagnosed with multicentric CD (MCD) and commenced siltuximab with ongoing reviews by the haematologists.
DISCUSSION
CD refers to a rare group of disorders with various aetiologies, presentations and management options, however, all presenting with lymph node enlargement and classical histopathological findings [1]. The disorders can be divided into unicentric Castleman disease (UCD), which involves a single lymph node or single area of lymph nodes, or MCD, which involves multiple lymph nodes [1]. MCD can be further divided based on aetiology to HHV8-MCD, POEMS-associated MCD or idiopathic MCD (iMCD) [1].
There are no known risk factors for UCD, POEMS-MCD and iMCD. On the other hand, HIV is the most common immunocompromised factor underlying HHV8-MCD, as well as country of origin, chronic viral hepatitis, organ transplant and consanguineous parents [1]. The pathogenesis of CD is poorly understood, however has been attributed to a proinflammatory hypercytokinaemia including IL-6 [2].
The disorders clinically manifest differently depending on their subtype. As UCD affects single or single regions of lymph nodes, patients are normally asymptomatic or mildly symptomatic [2]. On the other hand, MCD can present with constitutional symptoms such as fevers, night sweats, weight loss and fatigue, as well as splenomegaly [2].
Diagnosis requires an excised lymph node biopsy with findings consistent with CD histopathological characteristics [1]. In 2017, the Castleman Disease Collaborative Network (CDCN) published consensus diagnostic criteria for iMCD consisting of two major and minor criteria based on clinical presentation and laboratory features [3].
The CDCN subsequently published guidelines in 2018 detailing treatment of iMCD based on disease severity [4]. Non-severe iMCD should be treated with siltuximab, patients with limited symptomatology treated with rituximab and those with severe may require chemotherapy [4]. Further guidelines were published in 2020, which detailed that UCD may be treated with surgical excision that is usually curative of symptoms; however, in cases of unresectable UCD, asymptomatic cases may be observed, or symptomatic cases treated with therapies such as rituximab or steroids [4].
The incidence of CD is rare, with 1000–1500 cases of iMCD and 5000–6000 cases of UCD in the United States per year [4, 5]. CD in the porta hepatis is rare presentation, with few reports in the literature of such [6–10].
CD is also an important differential diagnosis to consider in cases of splenomegaly. MCD often manifests as widespread lymphadenopathy in the abdomen, pelvis, mediastinum, axilla, neck and inguinal regions [11]. Splenomegaly is also common systemic feature, with reports of 30–72% of MCD cases [12]. The presence of splenomegaly has been suggested to be a significant negative impact on prognosis of MCD patients [12].
The case highlights the difficulty in diagnosing CD despite multiple investigations and highlights the importance of considering CD as a rare differential diagnosis in cases of lymphadenopathy and splenomegaly. Although biopsies from more peripheral sites are preferred as surgical biopsies have the disadvantage of requiring general anaesthesia and are more invasive, laparoscopic excisional biopsy of a porta hepatis lymph node can be achieved to reach a definitive diagnosis.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.
DATA AVAILABILITY
No new data were created or analysed in this study.

{kind=link}