





Abstract
Susac syndrome (SS) is a rare microangiopathy affecting the precapillary arterioles of the brain, inner ear, and retina. We present a novel case of SS, presenting as acute incomplete bitemporal field loss in addition to temporally spaced neurological and vestibulocochlear symptoms. A 39-year-old female was referred to the ophthalmology clinic with acute incomplete bitemporal hemianopia and worsening hemicrania. History revealed progressive hearing loss, subjective short-term memory impairment, and vertigo temporally spaced over the preceding 12 months. Magnetic resonance brain revealed multiple small colosal lesions and liner ‘spoke’ lesions. Fundus fluorescein angiography revealed multiple branch retinal artery occlusions in the right eye. Audiometry confirmed bilateral sensorineural hearing loss. Treatment included intravenous corticosteroids and rituximab. This case highlights the importance of early consideration and evaluation of SS in individuals presenting with atypical ocular disturbances, where no clear cause can be elicited, in order to limit the sequelae of disease.
Introduction
Susac syndrome (SS) is a rare microangiopathy affecting the precapillary arterioles of the brain, inner ear, and retina [1–5]. SS typically affects young females aged 20–40 years [1, 2]. Current literature has described over 400 cases worldwide [2]. The disease is characterized by a clinical triad of encephalopathy, sensorineural hearing loss, and branch retinal artery occlusion (BRAO). The degree of involvement of all three systems varies, as each can develop as single events over a prolonged course [5].
Encephalopathy present in SS can manifest as headaches, motor/sensory disturbances, behaviour changes, cognitive impairment, and aphasia [2–5]. Hearing loss is often bilateral and may be associated with vestibulocochlear symptoms [4,5]. Typical audiometry illustrates sensorineural hearing loss of low or mid-tone frequencies [1–5]. Visual disturbances manifest as scotomas, black spots, and changes in visual acuity and can be unilateral or bilateral [4, 5]. As per the most recent criteria proposed by the European Susac Consortium, definite diagnosis can only be made by involvement of all three aforementioned systems [4]. Here, we present a novel Australian case of SS, presenting as acute incomplete bitemporal field loss in addition to temporally spaced neurological and vestibulocochlear symptoms.
Case presentation
A 39-year-old Caucasian female was referred to the ophthalmology clinic from the emergency department with sudden onset bitemporal hemianopia. Her medical history was significant for typical migraines. There was no previous ocular history. Family medical history included Factor V Leiden and Protein C deficiency. General and neurological examination was unremarkable. CT brain revealed a thin rim of pituitary tissue along the floor of the sella turcica suggesting a partial empty sella and possible idiopathic intracranial hypertension (IIH). A provisional diagnosis of migraine with aura was made and referral to the ophthalmology clinic.
In the ophthalmology clinic, intraocular pressure was within normal limits and best corrected visual acuity was 6/6 bilaterally. Optical Coherence Tomography (OCT) revealed normal retinal anatomy bilaterally. Humphrey 24–2 visual field testing demonstrated enlarged blind spots bilaterally, suggestive of temporal field defects (Fig. 1). Anterior segment and posterior segment examination was normal, with no evidence of tilted or swollen discs bilaterally. A Magentic resonance (MR) brain with venogram showed no abnormality or thrombosis. Consequently, a referral to neurology was made for further assessment and investigations.

Humphrey 24–2 visual field illustrating enlarged blind spots with evidence of early superonasal loss bilaterally.
Neurology review 6 months later revealed the patient’s headaches had progressively evolved from sporadic and migrainous in nature to continuous daily headaches over the course of 3 years. The scotoma persisted in the absence of these headaches. The enlarged blind spot despite the absence of papilloedema raised concerns about IIH. A lumbar puncture revealed an opening pressure of 22.5 cm H2O. Cellular analysis of cerebral spinal fluid showed an elevated protein and albumin of 1300 and 1100 mg/L respectively. White cells were elevated at 11 × 109 cells per litre, with normal glucose. Oligoclonal bands IgG were also present; however, these were not paired with a serum sample. Coagulation and vasculitic studies were all normal.
Subsequent neurology review unveiled progressive worsening of headaches and visual disturbances. She reported gradual deterioration in hearing and intermittent tinnitus preceding her visual symptoms, as well as episodic vertigo, intermittent word finding difficulties, and subjective short-term memory impairment. The combination of visual field defects, neurological symptoms, and hearing loss raised speculation for SS.
Repeat MRI at this time demonstrated several small T2 hyperintense foci within the deep white matter tracts of both cerebral hemispheres, the corpus callosum, and the thalamic and basal ganglion in addition to the typical linear ‘spoke’ lesions within the roof of the corpus callosum (Fig. 2). Fluorescein fundus angiography demonstrated multiple BRAOs and arterial wall hyperfluorescence (AWH) in the infratemporal region of the right eye, while the examination of the left eye remained normal (Fig. 3).
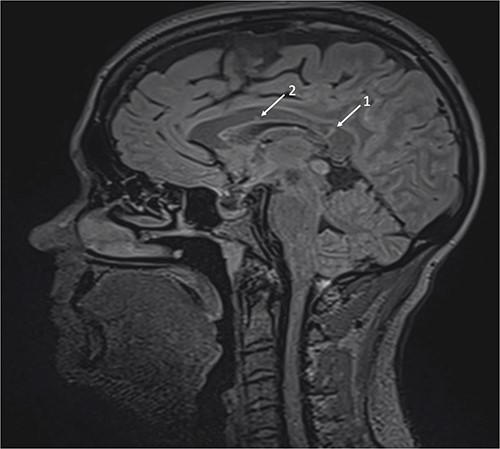
Brain magnetic resonance imaging (MRI) with sagittal T2 FLAIR illustrating linear ‘spoke’ (1) and T2 hyperintense lesions (2) in the corpus callosum.
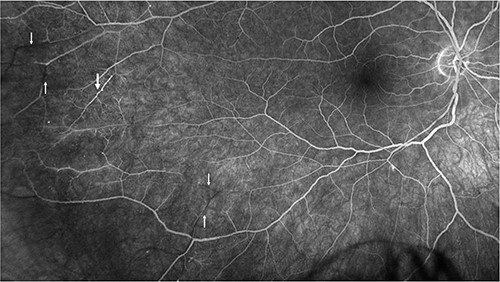
Fluorescein angiography of the right eye illustrating pathognomonic findings of BRAOs (thin arrows) and AWH (thick arrow).
Admission was arranged for further assessment and commencement of treatment based on presumption of SS. Audiometry illustrated mild sensorineural hearing loss to low frequencies (250–1000 Hz) in both ears. Immediate induction therapy of methylprednisolone 1 g intravenously for 3 days and intravenous immunoglobulins loading for a further 5 days was commenced. One month review revealed progressive reduction in the patients hearing, and hence a decision was made to incorporate rituximab for refractory SS.
Discussion and conclusions
SS presents as an inconspicuous diagnostic dilemma as the classic triad of symptoms generally develops as single events over a prolonged course, as opposed to only 13% of cases where symptoms present simultaneously [4]. Therefore, diagnosis is often delayed, which leads to protracted commencement of treatment and poorer outcomes [4, 5]. In our case, the identification of symptoms was temporally spaced, consequently resulting in a delayed diagnosis and the progression of symptoms.
The initial presentation of SS in our patient was characterized by bitemporal field deficits that did not conform to the vertical meridian. A review conducted by Heng et al. reported that encephalopathy was present in 72% of diagnosed SS patients, while visual disturbances were observed in only 24% of cases [5]. Furthermore, Kleffner et al. demonstrated that among 96% of patients meeting the retinal criteria for SS, 58% exhibited visual field defects during clinical investigation [4]. In our patient, intraretinal pathology was evident in the right eye, manifesting as multiple BRAOs and AWH, while the left eye did not exhibit significant retinal findings that correlated with the observed field deficits. Moreover, OCT revealed normal ganglion cell layer bilaterally, and no midchiasmal lesions or craniopharyngeal masses were detected, making compressive pathology an unlikely explanation in our patient.
In view of the diagnostic ambiguity, MR brain and audiometry were essential for definite diagnosis of SS. Repeat MR brain 8 months later exhibited extensive lesions consistent with SS and a progressive disease course. This illustrates that although neuroimaging is an important diagnostic criteria, lack of findings should not rule out the possibility of SS, as typical features can take several months to emerge [4, 5].
The clinical course and outcomes of SS are poorly understood, but immediate commencement of therapy should reduce disease-related sequelae [1]. No randomized control trials have investigated the treatment options for SS, hence empirical therapy is based on the autoimmune inflammatory aetiology [4]. In our case, the patient required monoclonal antibody treatment for refractory SS after 1 month of unsuccessful treatment on oral and intravenous steroids. This case highlights the importance of early consideration and evaluation of SS in individuals presenting with atypical ocular disturbances, where no clear cause can be elicited, in order to limit the sequelae of disease.
Author contributions
C. Gunaratnam., T. King., T. Moloney., M. Boggild., and T. Goodwin were all involved in the patients care, drafted the manuscript. All authors have read and approved the manuscript.
Conflict of interest statement
None declared.
Funding
No funding was obtained for this study.
Consent for publication
Written consent for publication was obtained from the patient.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.

{kind=link}
{kind=link}
{kind=link}