





Abstract
Primary adrenal pleomorphic spindle cell sarcoma (PSCS) is an exceedingly rare mesenchymal tumour that was previously known as malignant fibrous histiocytoma. It commonly occurs in extremities, retroperitoneum, peritoneal cavity and rarely in visceral organs. We report the first case of PSCS in the left adrenal gland in a 65-year-old female who presented with a large abdominal mass with vague symptoms. The wide range of differential diagnoses posed a challenge in establishing the diagnosis. However, this was overcome by appropriate radiological, intra-operative, histological and most importantly, comprehensive immunohistochemical findings. The patient underwent complete surgical resection of the tumour and had an unremarkable recovery. She remains without metastasis or recurrences to date through her 18 months of post-operative follow-up despite the poor prognosis of this tumour.
INTRODUCTION
Pleomorphic spindle cell sarcoma (PSCS), which was formerly known as malignant fibrous histiocytoma (MFH), arises from fibroblasts and histiocytes [1]. PSCSs are classified histologically into five groups, where storiform-pleomorphic type is the most common. There are few reports on metaplastic bone and cartilage formation within PSCS tumours [2]. The World Health Organization has classified PSCS as undifferentiated or unclassified sarcomas due to their unknown lineage. They are reported in extremities (67–75%), retroperitoneum (6–16%), peritoneal cavity (5–10%), rarely in abdominal viscera (liver, gall bladder and spleen) and are found exceedingly rarely in retroperitoneal organs (kidneys) [3, 4]. To the best of our knowledge, no cases of primary adrenal PSCS were reported.
PSCS represents a rare subgroup of adrenal cortical tumours named mesenchymal and stromal tumours. Primary adrenal PSCS is a diagnosis of exclusion due to multiple differential diagnoses for tumours in the area near the adrenal gland and the pathology of sarcoma and adrenal tumours. Therefore, clinical, radiological, histological and, more importantly, immunohistochemical markers play a crucial role in diagnosis. We report here the first case of a primary adrenal PSCS with focal osseous metaplasia, which presented as a vague large abdominal mass.
CASE REPORT
A 65-year-old female presented with vague left lower abdominal pain, back pain, abdominal fullness, lethargy and a non-ballotable lump at the left lumbar and iliac region for the past 3 months. She denied any genitourinary, gastrointestinal or gynaecological symptoms. Her investigations revealed microcytic-hypochromic anaemia, an erythrocyte sedimentation rate of 92 mm in the first hour and C-reactive protein of 24 mg/l. No monoclonal bands were present in the serum protein electrophoresis. Her renal and liver profiles, 9 AM cortisol, testosterone, 24-hour urine metanephrine and vanillyl mandelic acid were all within normal limits. There were no significant findings from pan endoscopy.
An abdominal ultrasound (US) revealed a 12 × 12 cm solid mass from the left suprarenal area (Fig. 1). Contrasted computed tomography (CT) revealed a 17.5 × 11.5 cm enhancing lesion in the left suprarenal region, completely replacing the left adrenal gland. The left kidney was inferiorly displaced, and coarse calcification was seen at the lesion’s periphery without lymph node enlargement. The contralateral adrenal gland, retroperitoneal and peritoneal structures were normal (Figs 2 and 3). Screening for primary carcinomas presenting as adrenal deposits utilizing clinical examination, pan endoscopy and contrasted CT were all negative. At this point, our working diagnosis was non-secreting left pheochromocytoma, and elective laparoscopic left adrenalectomy was planned.
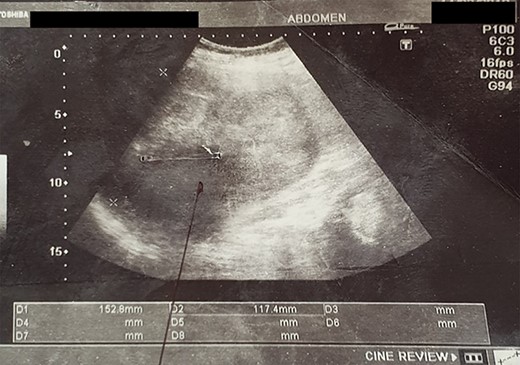
US image showing an altered echogenic left suprarenal lesion.
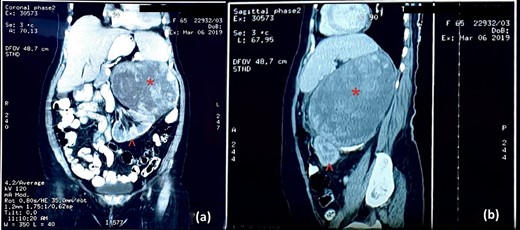
CT image: (a) coronal section and (b) sagittal section; the red asterisk mark in (a, b) shows the tumour and the red arrowhead in (a, b) shows the inferiorly displaced left kidney.
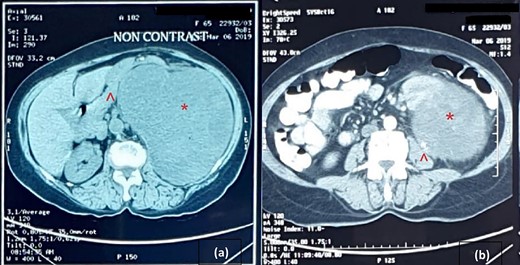
Cross-sectional image of computer tomogram; (a) non-contrasted image and (b) contrasted image; the red asterisk mark in (a, b) shows the tumour, the red arrowhead in (a) shows anteriorly displaced pancreas and the red arrowhead in (b) shows the clear distinction of tumour from the left psoas.
Unfortunately, she presented with rapid enlargement of the lump and worsening previous symptoms before the planned surgery. An urgent US examination showed an increase in the size of the tumour to 20 × 18 cm and therefore open left adrenalectomy was performed. At the time of surgery, we found a large well-defined 25 × 17cm capsulated mass in the left suprarenal region (Fig. 4) that was inferiorly displacing the left kidney and intestine. We could not identify the left adrenal gland. Complete resection of the tumour was performed, and she was discharged on the third post-operative day following unremarkable recovery.
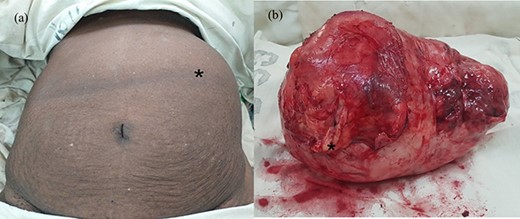
(a) Abdomen of the patient after induction at operating room and (b) macroscopic appearance of the tumour with the thick capsule; the black asterisk mark in (a) shows the tumour occupying the left abdomen and in (b) depicts an area of thickening on the capsule corresponding to osseous metaplasia.
Histopathological evaluation revealed a 24 × 21 × 15 cm tumour weighing 2.645 kg (Fig. 4b) with fibrotic, solid and cystic degenerative changes without capsular infiltration or vascular invasion. There were diffuse sheets of storiform-spindle cells with variable cellularity, bizarre multilobulated and bare nuclei, eosinophilic inclusions and high mitotic activity (Fig. 5). Foci of bony metaplasia were noted in the hard area of the capsule. No adrenal tissues were identified in the entire specimen. The tumour stained immunohistochemically positive for CD68, CD99, CD34 (40%), desmin (20%) and smooth muscle actin (SMA) (10%) but was negative for Pan-CK, S100, Melan A, epithelial membrane antigen (EMA), human melanoma black 45 (HMB45), CD117, B cell lymphoma 2, chromogranin and synaptophysin (Fig. 6). No metastases were identified on screening. We referred the patient to oncology for evaluation, but they did not recommend chemotherapy, and there have been no recurrences through the 18-month post-operative period.
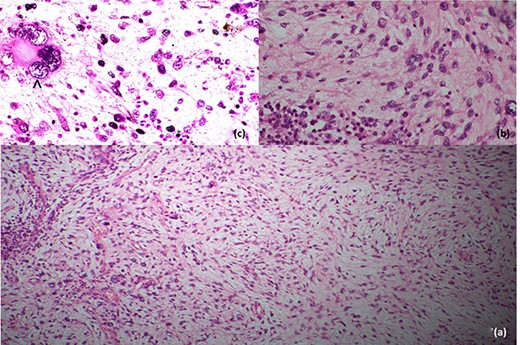
Histopathological features; (a) low-power image showing spindle cells in pleomorphic-storiform pattern, (b) high-power image showing spindle cells with bizarre multilobulated nuclei and (c) very high-power image depicting a nucleus in a spindle cell; the black arrowhead in (c) shows high mitotic activity in a spindle cell.
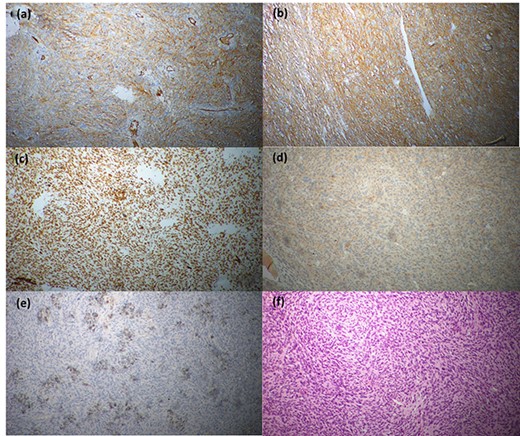
Immunohistochemical staining profile; (a) CD68-positive, (b) CD99-positive, (c) CD34-positive, (d) desmin-positive, (e) SMA-positive and (f) CD117-negative.
DISCUSSION
Adrenal tumours are mostly asymptomatic or present with abdominal lumps with nonspecific symptoms, similar to our patient [5]. Among the spectrum of adrenal tumours, primary mesenchymal tumours are exceedingly rare. Primary leiomyosarcomas are the most common malignant type [6]. At the time of presentation, our differential diagnoses included primary retroperitoneal sarcoma, metastatic tumours, liposarcoma, leiomyosarcoma, spindle cell carcinoma, malignant peripheral nerve sheath tumours, gastrointestinal stromal tumours, malignant melanoma, synovial sarcoma and pheochromocytoma [6].
Though PSCS can occur from any body part, they mainly originate from primitive mesenchymal cells [3]. PSCS has only been reported in extremities, abdominal and retroperitoneal viscera in the literature but not in adrenal tissue. The rarity of PSCS and the absence of risk factors of PSCS, such as radiation, trauma, chronic operative repair, surgical incisions and burn scars, in addition to no prior reported cases, suggested that primary adrenal PSCS is a less likely differential diagnosis in this case [7]. Bhagavan et al. reported five cases of MFH in extremities and pelvis with metaplastic bone and cartilage formation, similar to the finding of osseous metaplasia in this case [2]. The uncommon occurrence of metaplastic bone and cartilage in PSCS present a particular diagnostic problem in differentiating them from soft tissue osteogenic and chondrosarcomas, malignant mesenchymoma and giant cell tumour [2].
Detailed immunohistochemistry is needed to establish the diagnosis of PSCS of the adrenal gland. PSCS are variably positive for CD68, CD34 and CD99. Gastrointestinal stromal tumour, malignant peripheral nerve sheath tumour, malignant melanoma, synovial sarcoma, spindle cell carcinoma and pheochromocytoma are positive for CD117, S100, HMB45, cytokeratin, EMA, synaptophysin and chromogranin, respectively. Leiomyosarcoma is strongly positive for desmin, vimentin and SMA [8].
Surgical resection with free margin is the mainstay of treatment for PSCS, whereas radiotherapy, chemotherapy and immunotherapy are also described. Tumour size, location, site and histological types are the major determinants of prognosis in PSCS [9]. PSCS is an aggressive tumour with high recurrence and metastasis despite complete resection. A long-term follow-up with imaging is needed to look for recurrence and metastasis.
In conclusion, we report the first case of primary adrenal PSCS, which was initially investigated as a pheochromocytoma. We emphasize the importance of considering PSCS in the differential diagnosis for a large adrenal tumour along with other types of sarcomas. Establishing the diagnosis and evaluating the prognosis of PSCS should always encompass radiological, histological and full battery of immunohistochemical studies. PSCS carries a worrisome prognosis despite complete surgical resection, which will require close follow-up care.
ACKNOWLEDGEMENTS
The authors wish to thank Dr Benjamin Flink, MD, MPH, Bariatric and General Surgeon at Northside Hospital in Atlanta, GA, USA, and Dr OMO. Siddiqa, Registrar in Chemical Pathology at National hospital of Sri Lanka, Colombo, for their assistance in language editing, typesetting, formatting and image editing of this manuscript.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
No funds of any sort received by the authors for this article.
CONSENT
Informed written consent was obtained from the patient for publication of images and manuscript and is available for review at the unit.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}