





Abstract
Multiple primary cancers are defined as primary malignant tumors of different histological origins in one person. Desmoid-type fibromatosis (DF) is an extremely rare, locally aggressive, connective tissue malignancy that can be rooted anywhere in the body with the most common sites being thoracic wall and extremities. In contrast, granular cell tumors are rare neoplasms derived from Schwann cells commonly found in the oral cavity, skin and gastrointestinal tract. Moreover, diagnosing a patient with two primary cell tumors has become more common and the challenge of treatment becomes the focus in clinical situations. However, findings of a mass containing DF encapsulated by a granular cell tumor. Currently, there are no established guidelines for this rare condition. This case report serves to raise awareness of these two uncommon primary tumors emerging in an idiosyncratic nature.
INTRODUCTION
Desmoid-type fibromatosis (DF) is an extremely rare, locally aggressive, connective tissue malignancy that can be rooted anywhere in the body with the most common sites being thoracic wall and extremities. It has a reported incidence in 2–4 per million population and accounts for 0.03% of all neoplasms [1]. Although this tumor is associated with high rates of local recurrence, it lacks the potential to metastasize [2]. In accordance with recent literature, excision is no longer the primary method of treatment and initial surveillance is indicated to monitor the stabilization, progression or even recession of desmoid tumors. In contrast, granular cell tumors are rare neoplasms derived from Schwann cells commonly found in the oral cavity, skin and gastrointestinal tract that must be removed to avoid recurrence or metastasis [3, 4]. Granular cell tumors predominantly arise in women in the fourth to sixth decade with the overall incidence of 0.03% [3]. Moreover, diagnosing a patient with two primary cell tumors has become more common and the challenge of treatment becomes the focus in clinical situations. The overall frequency of multiple primaries ranges from 2 to 17% with a vast improvement in prevention, diagnosis and treatment [5]. In this case, however, findings of a mass containing DF encapsulated by a granular cell tumor, although better understood with improved diagnostic techniques, is an enigma that requires further discussion. This case report serves to raise awareness of these two uncommon primary tumors emerging in an idiosyncratic nature.
CASE REPORT
A 57-year-old female presents with a 3-year history of an enlarging right posterior neck mass. The mass was immobile and hard in nature. The patient reported the mass in 2019 with signs of progressive enlargement over the past several years. There was no reported drainage or signs of infection associated with the mass. The patient also denied any procedures or past enlargements in the region. After thorough discussion, the patient opted for an elective excision and consented for mass removal. The mass was excised to its entirety and sent for pathology examination. During the operation, a penrose drain was left in place. The following day in the clinic, the penrose drain was removed and the patient did not present with any postoperative complications.
Per gross report, the mass measured 3.5 cm. The mass was biopsied, which showed an infiltrative tumor involving skeletal muscle and composed of large, epithelioid cells with abundant, granular cytoplasm (Fig. 1), indicating the typical features of granular cell tumor. In the central aspect of the mass, a proliferation of monomorphic myofibroblast-like spindle cells in streaming fascicles were found (Fig. 1), also indicating the typical features of desmoid-type fibromatosis. Additionally, the granular cells showed pale basophilic to amphophilic cytoplasm. No pleomorphism was noted. The microscopic examination of the excised mass revealed various morphological tumor cells. Immunohistochemical stains showed that the epithelioid cells are positive for S100 (Fig. 2) and CD68 and negative for pan-cytokeratin. The spindle cells are focally positive for SMA in a myofibroblastic pattern.
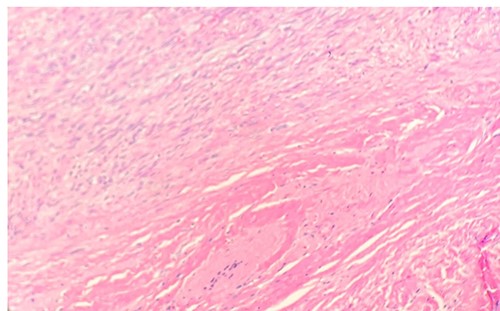
Microscopic examination reveals long sweeping fascicles with uniform elongated, slender, spindled cells with pale cytoplasm in a collagenous stroma (left upper- desmoid-type fibromatosis). Sheets of uniform epithelioid cells with abundant eosinophilic granular cytoplasm and small nuclei (right lower-granular cell tumor) (H&E 10x).
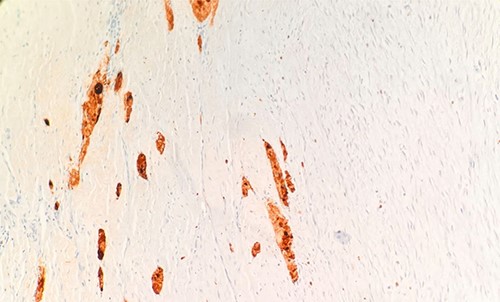
Microscopic examination reveals tumor cells are positive for S100. IHC 20X.
DISCUSSION
Multiple primary cancers are defined as primary malignant tumors of different histological origins in one person. Recently, there has been an increase in diagnosis of masses composed of multiple primary tumors due to improvements in diagnostic technology, increase in the longevity of patient survival and our understanding of malignancy [6]. Desmoid-type fibromatosis is an uncommon histologically benign disease, characterized by a monoclonal expansion of fibroblastic tissue [7]. Diagnosis is based on pathology, either via core biopsy or surgical excision of tissue. B-catenin gene mutation is thought to play a role in the pathophysiology of DF, as nuclear positivity is found in approximately 80% of sporadic DFs. However, screening for mutational analysis of B-catenin gene has not previously been shown to be a part of diagnostic workup for DF [8]. Although most cases of DF are sporadic, it has been strongly linked to populations exhibiting familial adenomatous polyposis (FAP) in ~5–10% of DF cases [9, 10]. A patient with FAP is at an 852-fold increased risk of developing a DF when compared to the general population [11]. Recent literature recommends nonsurgical treatments for DF due to their inability to metastasize. Noncytotoxic drugs, such as nonsteroidal anti-inflammatory drugs, and anti-estrogen drugs have been proven to reduce tumor size and symptoms in DF patients. Tamoxifen 30 mg daily has shown ~50% improvement in patients, whereas higher doses of tamoxifen (120–200 mg/day) has shown an even greater effect in 63–77% of patients [12]. The DF response to tamoxifen is gradual and must be evaluated after 6–12 months. Biological agents along with traditional chemotherapy drugs are also a part of medical management for DF. The management for DF is a stark contrast to the management of a granular cell tumor.
Granular cell tumor is a neoplasm, most commonly seen as benign cases, which arises in the skin, oral cavity or digestive tract. It is thought to originate from Schwann cells as it stains positive for both S-100 and neuron specific enolase. Granular cell tumors commonly target females more than males and age groups of 10–50 years old [13]. There is no role for medical management due to the low therapeutic value of chemotherapy. Therefore, complete resection of the mass with clear margins is the treatment for granular cell tumors, irrespective of malignancy. If the tumor is malignant, additional lymph node dissection is recommended. The prognosis after surgery is good with low recurrence rate [14].
Our patient’s right neck mass had a medium to high index of clinical suspicion to be cancerous due to the progressive enlargement and firmness. This case is interesting due to the idiosyncrasy of our patient’s neck mass being a desmoid-type fibromatosis encapsulating a granular cell tumor, which are two conditions with not only conflicting treatments but also have never been observed together in previous literature. Currently, there are no established guidelines for this rare condition. Periodic evaluations are recommended to monitor recurrency and metastatic spread.
Due to guidelines of granular cell tumors indicating complete resection with clear margins, the treatment of this mass involving both pathologies was sought to be the same. Close observation and performance of biopsy are indicated to rule out malignancy. This could ensure early diagnosis and treatment of a potentially life-threatening metastasis.
CONFLICT OF INTEREST STATEMENT
There are no conflicts to be declared.
FUNDING
None.

{kind=link}
{kind=link}