





Abstract
Sclerosing angiomatoid nodular transformation (SANT) of the spleen is a rare benign vascular lesion with unknown pathogenesis and no definitive pathognomonic radiological features. The majority of patients with SANT are asymptomatic, and the lesion is an incidental finding on cross-sectional imaging performed for unrelated reasons or during intra-abdominal surgery. However, in the symptomatic minority, abdominal pain is the most commonly reported symptom. SANT generally remains stable or has very slow growth, making it amenable to surveillance using serial cross-sectional imaging. Herein, we report the unusual case of SANT in a 30-year-old female with rapid growth from 6.0 × 5.6 × 4.4 cm to 8.0 × 6.6 × 7.2 cm over 21 months. Given the rapid growth, it was imperative to rule out malignancy. Thus, the patient underwent a laparoscopic total splenectomy. For SANT, splenectomy serves the dual purpose of diagnosis and definitive therapy.
INTRODUCTION
Sclerosing angiomatoid nodular transformation (SANT) of the spleen is an extremely rare benign vascular lesion that was first reported in a case series of 25 patients [1–8]. While its pathogenesis remains a subject of much debate, some consider it as a manifestation of reactive inflammatory disease linked to immunoglobulin G4 (IgG4)-related sclerosing disease [3, 5]. Interestingly, in the majority of cases, SANT remains stable or has very slow growth, making it amenable to surveillance using serial imaging. Herein, we report a rare case of rapidly growing SANT of the spleen in a 30-year-old female, concerning for angiosarcoma.
CASE PRESENTATION
A 30-year-old female presented with a 6-month history of intermittent, dull and non-radiating epigastric pain. Her medical history was significant for idiopathic intracranial hypertension. Patient denied a history of smoking, alcohol or illicit drug use. Family history was significant for diabetes mellitus and hypertension. Vital signs and all systemic examinations were unremarkable.
Laboratory investigations, including complete blood count, electrolytes and liver function tests, were within normal limits.
Abdominal ultrasonography showed a 5.0 × 5.0 cm well-defined, rounded, lobulated and hypoechoic vascular solid splenic lesion with no increased transmission or calcification. This lesion had vessels radiating from the center to the periphery (Fig. 1). Further evaluation with contrast-enhanced computed tomography (CT) of the abdomen showed a 6.0 × 5.6 × 4.4 cm solitary, rounded and lobulated non-calcified mass in the spleen, demonstrating heterogeneous, linear and nodular arterial enhancement with progressive filling in the portal venous and delayed phases (Fig. 2).
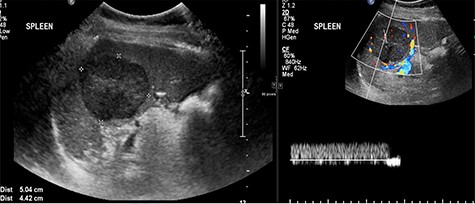
Ultrasound images with Doppler showing a well-defined rounded hypoechoic mass with vascularity in the peripheral areas as well as inside the lesion with no through transmission.
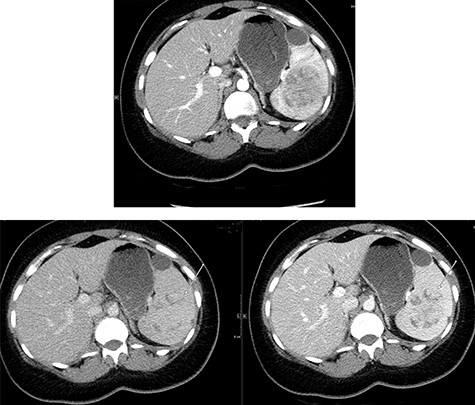
Top image shows abdominal CT scan arterial phase axial section with a hypodense, solitary, rounded and lobulated non-calcified mass lesion in spleen. Bottom right and left images show abdominal CT scan portovenous and delayed phase axial images showing a hypodense lesion and areas of filling (arrows in both images) which becomes mostly isodense to spleen; incidental finding of a splenic cyst.
A multi-planar, multi-sequential abdominal magnetic resonance imaging (MRI) without contrast showed a well-circumscribed splenic mass with patchy nodular enhancement, which became more diffuse on delayed images and had low T2 signal and isointense T1 signal with no diffusion restriction (Fig. 3). There was no evidence of lymphadenopathy and no features favoring a lymphoma. The splenic mass was diagnosed as SANT, with plans to follow it with serial imaging.
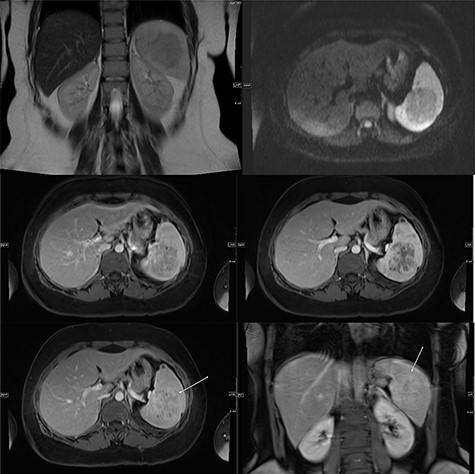
(Top) coronal T2 WI MR Image showing a hypointense lesion in the spleen, which is not showing any diffusion restriction (right image); (middle images) post-contrast axial T1 WI MR image showing hypointense lesion with patchy contrast enhancement in the center (middle right image); the lesion becomes further isointense on delayed image (middle left image); (bottom) MRI of abdomen T1 weighted axial (bottom left) and coronal (bottom right) post-contrast images showing a lesion in the spleen (arrow) with peripheral and septal enhancement with a hypoenhancing center.
A repeat contrast-enhanced CT scan 21 months later showed an increase in size of the lesion to 8.0 × 7.2 × 6.6 cm (Fig. 4). Positron emission tomography (PET) CT demonstrated the mass to be moderately hypermetabolic with a standardized uptake value (SUV) of 4.7 (Fig. 5).
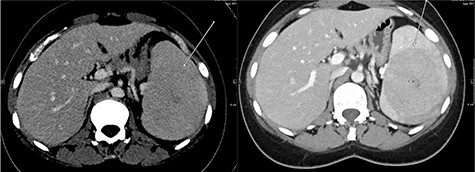
The post-contrast CT axial image showing central hypodense splenic lesion in portovenous phase (arrow—right image) which becomes isodense on delayed images (arrow—left image).
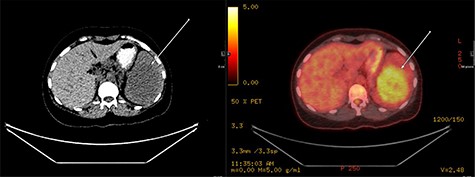
PET fused axial image with corresponding low-dose unenhanced CT image (left); the lesion is showing moderate FDG uptake (arrow).
The splenic mass was suspected to be SANT. However, the rapid growth was concerning for angiosarcoma. Given the rapid growth in size and the concern for angiosarcoma, the patient underwent a laparoscopic total splenectomy.
Histopathological analysis of the surgical specimen revealed a tan, fleshy and well-circumscribed lesion measuring 8.0 × 7.0 × 6.0 cm, with nodules surrounded by collagen fibers. These nodules had slit-like, irregular vascular spaces lined by plump endothelial cells with some spindle and ovoid cells. No nuclear atypia or mitoses were seen. On immunohistochemistry, CD31 (clone JC70A) stained all the endothelial cells, CD34 (Qbend10) stained only in the capillaries and CD8 (C8/144B) was positive in the sinusoids. The macrophages in the background were highlighted by CD68 (KP1) and CD163 (MRQ-26). These findings were consistent with SANT.
DISCUSSION
A male-to-female ratio of 1:2 and an age range of 3–82 years have been described in literature for SANT [2, 3]. The true incidence of SANT remains undetermined since the majority of patients with SANT are asymptomatic, and the lesion is an incidental finding on imaging performed for unrelated reasons or during intra-abdominal surgery [2–4]. However, in the symptomatic minority, abdominal pain (in 25.8%) is the most commonly reported symptom [3]. Other signs and symptoms that have been described in literature include pelvic or flank pain, vomiting, constitutional symptoms (such as fever, weight loss and night sweats), hematologic abnormalities (e.g. thrombocytopenia, pancytopenia and anemia) and splenomegaly [2, 3, 7].
The pathogenesis of SANT remains a subject of much controversy [8]. Some of the commonly described hypotheses on its pathogenesis include an abnormal harmartomatous transformation of splenic red pulp, resulting from an excessive non-neoplastic stromal proliferation, Epstein–Barr virus association and IgG4-related disease, among others [1, 3, 4, 6, 8, 9]. SANT also has a wide range of differential diagnoses, including hamartoma, hemangioma, lymphangioma, inflammatory pseudo-tumor, angiosarcoma, malignant fibrous histiocytomas, pleomorphic undifferentiated sarcomas, lymphomas, hemangioendothelioma and metastases [3, 4, 7–9]. Differentiating SANT from these conditions, even with multiple imaging modalities, can present as diagnostic challenge; therefore, histopathology is the gold standard for diagnosis [4, 6].
While there are no established pathognomonic radiological features for SANT, a ‘spoke and wheel’ appearance, with enhancement of radiating lines and rim on contrast-enhanced CT or MRI has been widely reported [3, 10, 11]. Use of fluorodeoxyglucose (FDG) PET/CT to definitively diagnose SANT or rule out malignancy has proven challenging since radiological features generally characteristic of malignant lesions, such as high FDG uptake, have also been described with SANT [2, 6, 9, 10]. Image-guided core needle tissue sampling poses the risk of complications, such as splenic rupture, life-threatening hemorrhage and intraperitoneal dissemination in case of a malignant lesion [2, 3, 6, 11]. Thus, splenectomy, which serves the dual purpose of diagnosis and definitive therapy, is recommended when suspicion for malignancy is high [2–4, 6, 9].
The preferred management for SANT is surveillance via serial imaging, particularly when suspicion for malignancy is low. In the majority of cases, SANT is stable or slow-growing. However, rapid growth with SANT has been reported in the pediatric population [12, 13]. The rapid growth seen in our case, from 6.0 × 5.6 × 4.4 cm to 8.0 × 6.6 × 7.2 cm within just 21 months, in an adult is extremely rare. Our extensive literature review identified only one case of rapid growth in SANT in adults [8]. In this particular case, the rapid growth was presumed be the result of adrenalectomy and the concurrent fall in cortisol, which led to growth of the IgG4-related sclerosing disease-related SANT lesion. We recommend that although surveillance with serial imaging is the generally accepted management for lesions believed to be SANT, splenectomy should be performed to rule out malignancy where suspicion for malignancy is high, such as in cases with rapid growth.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}