





Abstract
Ganglioneuroma is a nerve tumor arising from the sympathetic neural crest. It is a rare benign tumor. Retroperitoneum is its second location after the posterior mediastinum. Usually asymptomatic, it is discovered incidentally on imaging. Surgical resection is the sole treatment. The prognosis is good if the diagnosis is made early with quality R0 surgical excision. We report a case in a 14-year-old female admitted to the emergency department for obstructive pyelonephritis. Imaging features found a retroperitoneal mass with characteristics suggestive of a retroperitoneal ganglioneuroma, which was confirmed by histological study. Ganglioneuroma should be a part of differential diagnoses for any retroperitoneal mass in children and young adults.
INTRODUCTION
Ganglioneuroma (GN) is a benign nerve tumor in children and young adults arising from the sympathetic neural crest. It is a rare, usually asymptomatic tumor. It is considered as a radiological incidentaloma with increasing incidence due to multiple imaging performed.
We report a case of GN revealed by obstructive pyelonephritis in a 14-year-old patient. We discuss radiological features and therapeutics strategies of this rare entity based on the recent literature.
CASE REPORT
A 14-year-old female patient with a history of intermittent right flank pains treated symptomatically without any radiological exploration was admitted to the emergency department for exacerbation of the pain associated with fever. Physical examination found a lumbar contact, guarding in the flank and a temperature of 40°C. Laboratory workup performed showed urinary tract infection, and parenteral antibiotic therapy based on ceftriaxone 2 g/day for 10 days was introduced.
Contrast computed tomography (CT) scan revealed a voluminous (173 × 67 × 59 mm), well-limited and lobulated, right retroperitoneal tumor, hypodense, with delayed and low enhancement (Fig. 1).
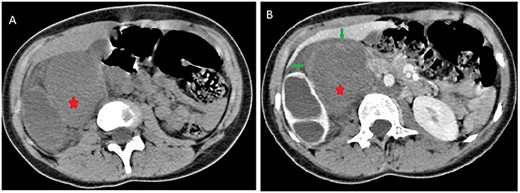
Abdominal CT scan showing a retroperitoneal mass; retroperitoneal ganglioneuroma (red spark) before (A) and after (B) heterogeneous enhancement (green arrow).
On magnetic resonance imaging (MRI), tumor signal was heterogeneously high in T2-weighted images, while iso to low in T1-weighted images delayed heterogeneous enhancement with some septa and areas of necrosis (Fig. 2).
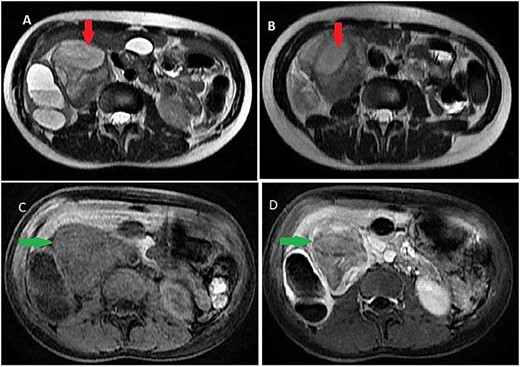
Abdominal MRI showing a retroperitoneal mass; (A and B) axial T2-weighted images show heterogeneous high signal in the lesion; (C and D) axial T1-weighted image, before (C) and after (D) dynamic sequences with contrast showing a delayed heterogeneous enhancement with some septa and areas of necrosis within the mass.
It was abutting the abdominal aorta, inferior vena cava and right renal pedicle. It extended toward the liver and right diaphragmatic pillar. It was compressing the kidney and ureter with ureterohydronephrosis and cortical thinning. Renal calyces and pelvis contents were dense and their walls enhanced (Fig. 3). The adrenal gland was normal.
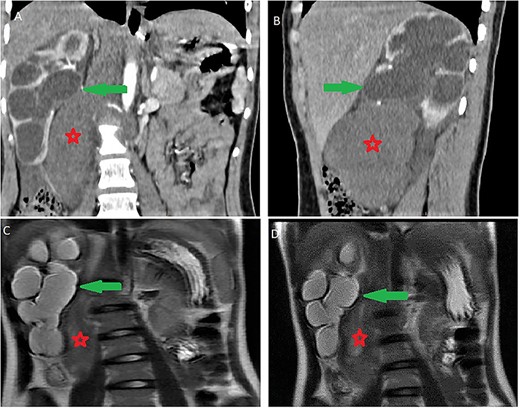
Ureterohydronephrosis due to the mass effect; ureterohydronephrosis (green arrow) due to the mass effect (red spark) on coronal (A), sagittal (B) Ct scan and coronal MRI T2-weighted image (C and D).
Based on this observation, obstructive pyelonephritis due to compression by GN was suspected. Other differential diagnoses of tumors, such as neuroblastoma, ganglioneuroblastoma, germinal tumor and paraganglioma, were also suspected.
Alpha-fetoprotein and methoxylated derivatives levels were normal. Subsequently, a CT-guided biopsy was performed to confirm the diagnosis of GN (Fig. 4).
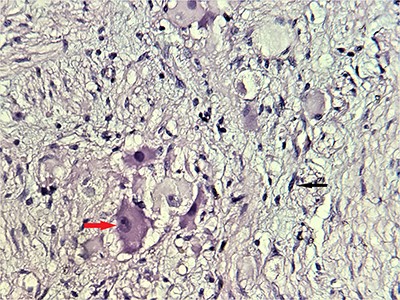
Microscopic scrutiny of the resected specimen; the observed ganglion cells (red arrow) are mature, having a compact, eosinophilic cytoplasm and a single eccentric nucleus with prominent nucleolus; Schwann cells (black arrow) are also mature—HE;400X.
A laparotomy surgical resection was performed, after a multi-disciplinary team decision. The post-operative was uneventful and renal function remained normal. She was discharged on day seven of post-operative.
Histologic work-up of the surgical specimen had confirmed the absence of malignancy especially an immature neuroblastic tissue.
A clinical 2 months follow-up after surgery was satisfactory.
DISCUSSION
GN is a rare benign tumor with an incidence in a population estimated to one per million person-years, and it constitutes only 0.72–1.6% of primary retroperitoneal tumor [1]. A higher frequency has been reported in patients with multiple endocrine neoplasia type II and neurofibromatosis type 1 [2].
GN is a well-differentiated tumor of the sympathetic nervous system, arising from the neural crest [3] which belongs to the spectrum of neuroblastic tumors, including neuroblastoma and ganglioneuroblastoma [4]. It can also come from the maturation of pre-existent neuroblastoma spontaneously or after chemotherapy [5]. It is composed of gangliocytes and mature stroma [6]. This tumor can grow at any site of the sympathetic nervous tissue and it is mainly distributed in the posterior mediastinum, retroperitoneum, neck and adrenal gland in descending order [7].
First described by Loretz in 1870, GN is mostly seen in pediatric populations, with 60% of cases before 20 years [8]. The median age of the diagnosis is ~7 years [4]. A female predominance with a sex ratio of 3:2 has been described [8].
GN is usually an incidental asymptomatic finding on abdominal imaging. Sometimes when its size increases, it can cause some non-specific symptoms due to the compression on neighboring organs, as was reported in our case. Some GN secretes catecholamine and vasoactive intestinal peptide causing hypertension, sweating and diarrhea.
CT and MRI are the best imaging techniques for the diagnosis and characterization of GN. On imaging, it appears as an oval, well-delineated mass and tends to grow around major blood vessels without narrowing them [3]. On CT, it is a hypodense mass with a density between 20 and 40 HU and delayed contrast enhancement in a range of 10–20 HU. GN appears homogeneous and can be sometimes heterogeneous with circumscribed or spotted calcifications (in 20% of the patients) [9]. On MRI, T1-weighted images show a low or intermediate signal intensity, whereas T2-weighted images show a heterogeneous intermediate or high-signal intensity. This variability of the signal on T2-weighted images is due to two factors: the ratio of myxoid stroma to cellularity and the amount of collagen present within the tumor [3]. MRI contrast enhancement is most often slight and heterogeneous [1].
There is no distinct pathognomonic radiological feature of GN, but the lack of enhancement is quite suggestive. Before the histologic study, clinical and radiological diagnosis is challenging with several differential diagnoses. Among these diagnoses, secreting paraganglioma must be eliminated before any biopsy by measuring methoxylated derivatives. A CT-guided biopsy can then be carried out to confirm the histological nature of the mass.
Histologically, it is characterized by large ganglions cells proliferation with eosinophilic cytoplasm, large clear nucleolus without atypia, in a loose fibrillar background. This was the case of our patient; no immature neuroblastic component was noted. Histological study of the surgical specimen is essential, to eliminate a contingent of neuroblastoma and pheochromocytoma within the GN, which can be missing on a core biopsy [10].
Resection is the sole treatment. Sometimes in a large tumor, surgery can be challenging because it tends to extent to neighboring anatomical vital structures. After complete resection, the prognosis is good. However, some cases of progression, late recurrence or malignant transformation have been reported, especially after incomplete resection [1]. Furthermore, a long-term follow-up including imaging control is necessary to prevent potential relapse. There is no need for neoadjuvant or adjuvant treatment [8].
In conclusion, retroperitoneal ganglioneuroma should be a part of differential diagnoses for any retroperitoneal mass in children and young adults. Its radiological appearance without being pathognomonic is quite suggestive and needs histologic confirmation. Surgery with a long-term clinical and mostly radiological follow-up is the sole treatment.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
All authors have declared that no financial support was received from any organization for the submitted work.
CONSENT
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
AUTHOR CONTRIBUTIONS
All the authors testified to the care of the patient and the writing of the manuscript. The authors have read and agreed with the contents of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}