





Abstract
Leiomyosarcoma (LMS) is a common form of soft tissue sarcoma. Primary colonic LMS is an extremely rare entity, comprising 1–2% of gastrointestinal malignancies. Primary mesenchymal sarcomas of the gastrointestinal system are rare and constitute just 0.1–3% of all gastrointestinal tumours. LMS is the most common variant of such tumours and represents just 0.12% of colorectal malignancies.
We present a case of a 65-year-old female, who presented to the emergency department with 3 days history of obstipation and generalized abdominal pain. Radiology (X-ray and ultrasound) indicated a large pelvic mass compressing the sigmoid colon and its surrounding structures.
Histopathological analysis indicated a primary LMS of the sigmoid colon. Diagnosis is established mostly postoperatively after histopathological evaluation. Prognosis and treatment modalities for this aggressive malignancy remain insufficient. LMS is relatively impervious to chemotherapy/radiotherapy. Our patient was treated by surgical excision of the tumour and referred postoperatively for adjuvant chemotherapy.
INTRODUCTION
Primary mesenchymal sarcomas of the gastrointestinal system are rare and make up merely 0.1–3% of all gastrointestinal tumours [1]. Leiomyosarcoma (LMS) is the most common variant of those tumours and constitutes just 0.12% of all colon malignancies [2]. Among the diversity of soft tissue sarcomas, LMSs represent 10–20% of those malignancies. LMS mostly arises in the uterus, GI tract and retroperitoneum. Within the gastrointestinal (GI) system, the stomach is the most common site followed by the small intestine, colon and rectum [3]. Scarce data are available about its molecular heterogeneity, and no targeted therapy so far is established for LMS [4]. The classical colon LMS presents mainly with an array of non-specific symptoms such as mild abdominal pain, fresh/obscure bleeding per rectum and weight loss.
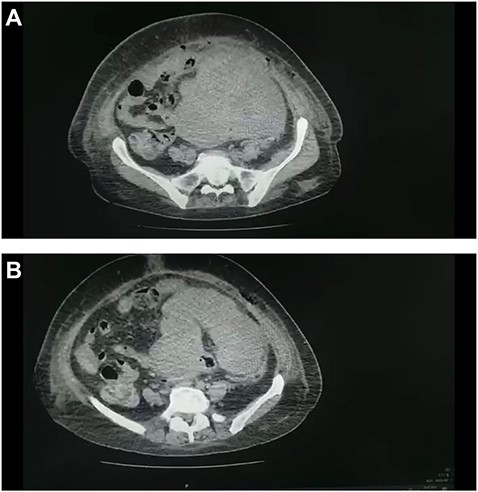
(A and B) CT scan of the abdomen and pelvis demonstrated a large pelvic mass adherent to the sigmoid colon and compressing its surrounding structures.
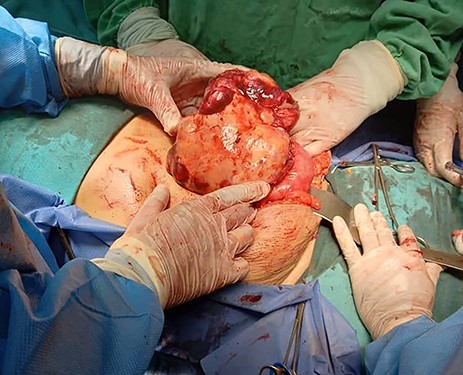
Tumour arising from the sigmoid colon, adherent to the small bowel and measured 12 × 13 × 16 cm.
CASE REPORT
We present the case of a type-2 diabetic 65-year-old female, who presented to the emergency department with 3-day history of obstipation and generalized abdominal pain accompanied by a gradual asymmetric distention of the abdomen, nausea and lossof appetite. No fever, vomiting, bleeding per rectum or genitourinary symptoms were reported. Radiology (X-ray, ultrasound) suggested a significant pelvic mass compressing the sigmoid colon and its surrounding structures. Negative family, drug and allergic histories.
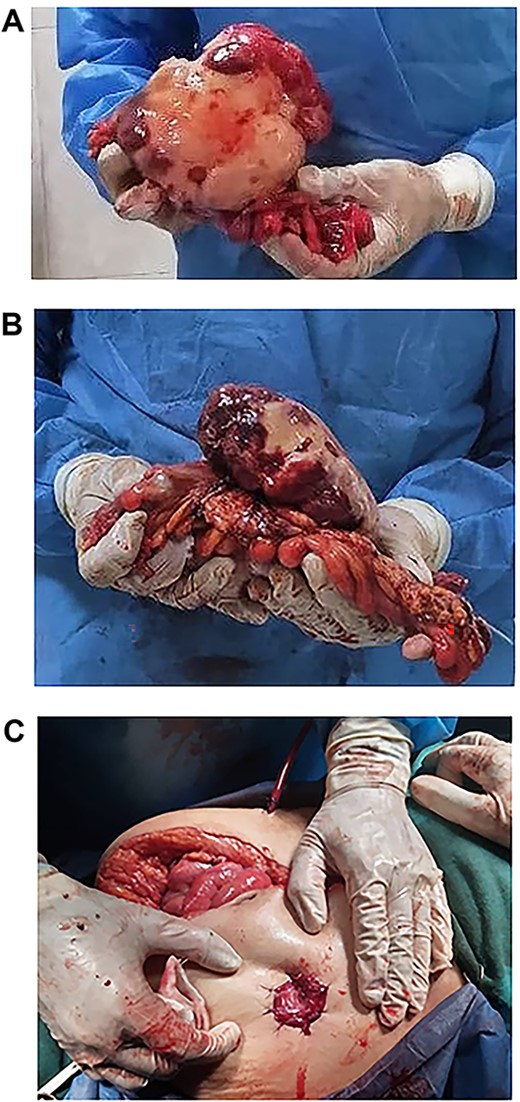
(A, B and C) Tumour completely excised along with the sigmoid colon and its mesentery, and an end colostomy was created.
Clinical examination revealed tachypnea and tachycardia, otherwise normal vital signs. Upon inspection, the abdomen was asymmetrically and significantly distended below and around the umbilicus. On palpation, there was generalized guarding and tenderness. A large hard mass with ill-defined borders was palpated. Bowel sounds were faint. Digital Rectal Exam (DRE) revealed traces of stool lacking visible blood.
Laboratory investigations were notable for anaemia and hypokalemia, otherwise within normal ranges.
Abdominal ultrasound revealed a bulky hypoechoic pelvic mass with ill-defined borders. Computed tomography (CT) scan of abdomen and pelvis established a large pelvic mass adherent to the sigmoid colon and compressing its surrounding organs measuring approximately 10 × 11 × 13 cm. (Fig. 1A and B). Initial management included intravenous fluid resuscitation, analgesics and prophylactic antibiotics.
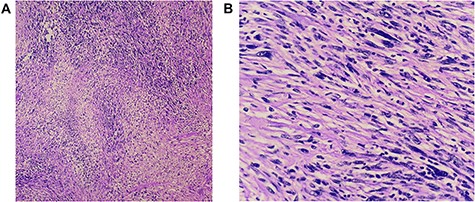
(A and B) Histopathology revealed a spindle cell neoplasm consistent with low-grade spindle cell sarcoma. There is no angioinvasion; border lines of resection are free of tumour.
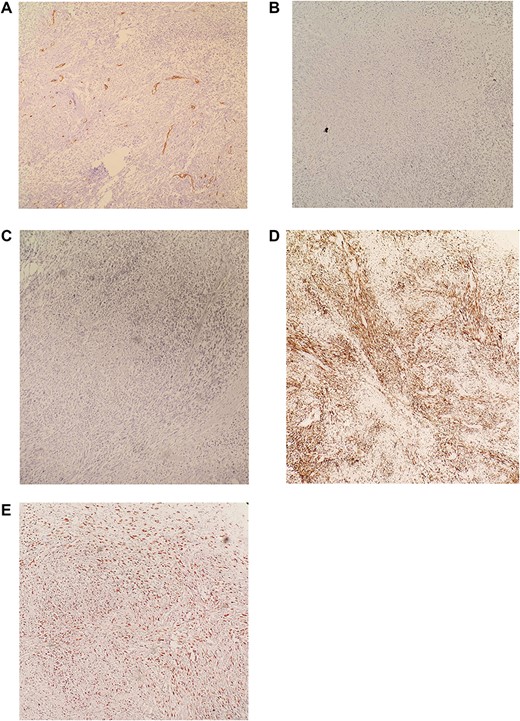
(A) IHC revealed CD34 negative. (B) IHC revealed CD117 negative. (C) IHC revealed S100 negative. (D) IHC revealed DESMIN positive. (E) IHC revealed KI-67 >30%. Diagnosis is consistent with moderately differentiated Leiomyosarcoma.
Exploratory laparotomy revealed a mass arising from the sigmoid colon, adherent to the small bowel and measuring 12 × 13 × 16 cm (Fig. 2). The tumour was isolated from its surroundings and completely excised in addition to a segmental bowel resection ‘en block’ for the sigmoid colon and its mesentery along with a 4 cm free margin, and the proximal end was created as an end colostomy (Hartmann’s procedure) (Fig. 3A, B and C).
Histopathology (hematoxylin and eosin) revealed a spindle cell neoplasm consistent with low-grade spindle cell sarcoma. No angioinvasion and lines of resection were free of tumour (Fig. 4A and B).
Immunohistochemistry (IHC) stained negative for (CD34-CD117-S100, and Vimentin) and stained positive for DESMIN while KI-67>30%. Diagnosis is consistent with moderately differentiated Leimyosarcoma (Fig. 5A–E).
The patient had successful recovery and has been followed up in the outpatient settings for 1 month following her operation and was referred to a specialized oncology hospital where she will receive adjuvant chemotherapy. She has no current active symptoms.
One-month post-operative computed tomography (CT) scan of the chest/abdomen/pelvis revealed: clear lung fields free of metastasis/lymphadenopathy, liver free of metastasis, no retroperitoneal/pelvic lymphadenopathy, no fluid levels near the surgical intervention field, no masses or signs of relapse/recurrence, and kidneys/spleen/genitourinary system are normal (Fig. 6A–D).
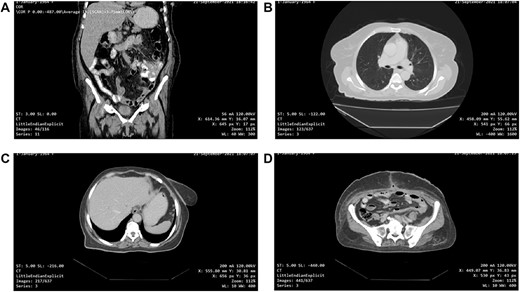
(A, B, C and D) One-month post-operative CT scan of the chest/abdomen/pelvis revealing a coronal view of the abdomen and pelvis/lung/liver/surgical site/pelvis free of tumour spread/metastasis/relapse.
DISCUSSION
The classical presentation of LMS of the gastrointestinal system is in middle-aged adults with an average age of diagnosis of 50 years [1]. In a series of 80 patients with various smooth-muscle tumours of the gastrointestinal tract, ‘Chou et al.’ [5] identified the most common presenting symptoms and signs as gastrointestinal bleeding (44%), abdominal mass (38%) and abdominal pain (21%). Surgical excision is considered the gold standard of treatment for soft tissue sarcomas and wide resections with clear margins are essential to limit local recurrence. Excised materials will then be sent for histopathological analysis to establish a diagnosis where LMS will positively stain for smooth muscle actin and desmin, and will show negative CD117, CD34 and DOG1 stains, which stain positive in GIST [2]. The most common cause of death in those patients is secondary to haematogenous spread of the tumour to the lung and liver [6]. In a study of 11 patients with colonic LMS by Aggarwal et al. [7] in 2012, just 2 of the 11 patients survived for 5 years, with a mean survival duration of twenty months. Yamamoto et al. [8] reported an approximated 5-year tumour-specific overall survival rate of 51.6%. Grade and size are the most paramount prognostic factors for disease-specific survival and later recurrence rate in patients with primary LMS [9]. Additional prognostic factors, which signify poor prognosis, include age >45 years, necrotic areas within the tumour, dissemination of disease and tumour size [1, 8].
LMSs arise primarily in the small intestine (45%) and colon (38%) and are genuinely rare in the stomach and oesophagus [8]. LMS comprised 57.5% of a series of 433 patients with primary colorectal sarcoma reported between 1998 and 2012 in the National Cancer Data Base, USA by Thiels et al [10]. Magnetic resonance imagings (MRI) are thus far the gold standard in the investigation for sarcomas [11]. In our case, MRI was unavailable. Immunohistochemical staining is the definitive mode of diagnosis. Characteristic features of LMS involved positivity for desmin, alpha-SMA, vimentin and H-caldesmon, and negativity for GIST markers: KIT, CD34, CD117 and DOG1 [12].
In conclusion, LMS of the colon is a rare, unpredictable and aggressive neoplasm with poor prognosis. It can mimic other tumours and GI illnesses but should be distinguished as a separate entity as the prognosis and treatment differ significantly. Nevertheless, it should be considered as a differential diagnosis when a patient presents with surgical abdomen. The prognosis of patients with LMS generally seems to be interrelated with the degree of histological grading. Site is not a vital independent prognostic factor for local recurrence in previous studies [13]. Surgery is the core treatment modality for primary colonic LMS, as most of the reported cases were diagnosed by histochemical staining of surgically excised specimens. LMS is relatively insensitive to chemotherapy [7]. Long-term follow-up of LMS patients is essential.
ACKNOWLEDGEMENTS
Chatti Lab: Histopathology lab responsible for analysing and staining the specimens excised during the surgical operation, Sibky Park, Arrawdah, Damascus, Syria.
Digital Scan and Interventional Radiology clinic: responsible for performing the post-operative CT scan of the patient and analysing it, Al- Mouwasat University Hospital Sq. Mazzah, Damascus, Syria.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}