





Abstract
Fibrous solitary tumors (FST) are mesenchymal tumors that can appear in different body regions. It is estimated that around 30% are found in the thoracic region, while rarely in meninges, abdomen, pelvis, extremities and bones. A correct diagnosis is important because 15–20% of cases develop a malignant behavior. Treatment of choice is surgical and posterior follow-up is essential. We present two atypical extrapleural FST cases, diagnosed in our center. Both were treated with surgery and in one case arterial embolization to reduce the bleeding risk was previously done.
INTRODUCTION
Fibrous solitary tumors (FST) are considered soft tissue mesenchymal tumors with a fibroblastic or myofibroblastic origin [1]. They were initially described as pleural tumors; however, there is literature describing them in many body regions. The most common location is in the pleura (between 30–80%) and less frequently in meninges and abdomen [2]. FST has an incidence of 2.8/100.000 population, with similar rates between men and women, and it is more frequent during the 50–60th decade. FST are usually asymptomatic with a slow growing behavior and in many cases, diagnosis is found incidentally in the image scans. In the abdomen, they can produce obstructive symptoms, mass effect or pain if size is big enough. Histologically, they use to have a prominent blood supply with a hemangiopericitic growth pattern. Immunohistochemistry (IHQ) shows CD34 and vimentin positivity. A correct diagnosis is very important as they are currently considered benign tumors, and surgery is the treatment of choice. Closer follow-up is strictly recommended if size is bigger than 10 cm or malignant histological component exists.
CASE REPORTS
This article presents two clinical cases of FST treated in our clinic. Both are extrapleural and with atypical location. The first was located in the prevesical space and the other in the superficial muscular aponeurosis. A review of the scientific literature is also done.
Case 1
A 43-year-old male patient was referred to hospital with micturition disorders and hypogastric abdominal pain during the previous few months. A computerized tomography (CT) was carried out showing a pre-vesical mass 6 × 5 cm in size with well-defined borders and hypercaptant during the arterial phase (Fig. 1). As an FST was suggested, a fine-needle aspiration (FNA) biopsy was done, but diagnosis was not confirmed.
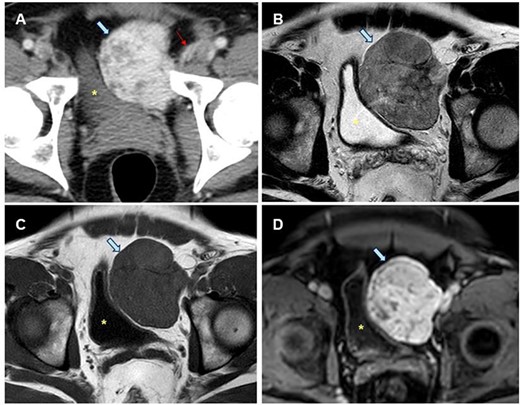
Radiological study of the pelvic mass. (A) CT scan with intravenous contrast: hypervascular mass (blue arrow). Epigastric vessels give vascular supply, coming from left common femoral artery (thin red arrow). (B, C, D). MRI in late T2-FSE (B), T1-FSE (C) and T1-FAT-SAT GD sequences. Hypointense mass both in T1 and T2 sequences without intravenous contrast suggesting the fibrous origin. It is confirmed in T1 sequence with fat saturation and after intravenous gadolinium administration is homogeneous, in late phase (8 min). Bladder is displaced towards the opposite side (yellow dot).
Radical surgery was decided after discussion in the tumors multidisciplinary committee, and previous vascular embolization was proposed to reduce the potential risk of bleeding (Fig. 2).
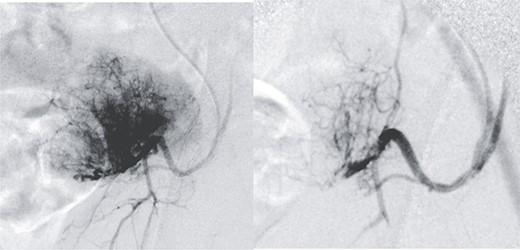
Presurgical tumor embolization. (A) By contralateral approach, left femoral diagnostic angiography is performed, showing hypervascular tumor pattern depending on the left epigastric artery. (B) Control after embolization of the two medial thirds of the tumor with calibrated particles of 600 +/− 75 microns. The rest of the tumor cannot be safely embolized due to the existence of obturator branches.
Laparoscopic approach is always an option but, in this case, to remove the tumor, we planned a Pfannenstiel incision. The decision was made due to the localization of the tumor.
During the surgery, the mass was identified at pre-vesical region (extra peritoneum) firmly attached to the pubis, in vicinity with the bladder, displacing it but without infiltration, so bladder resection was not needed (Fig. 3).
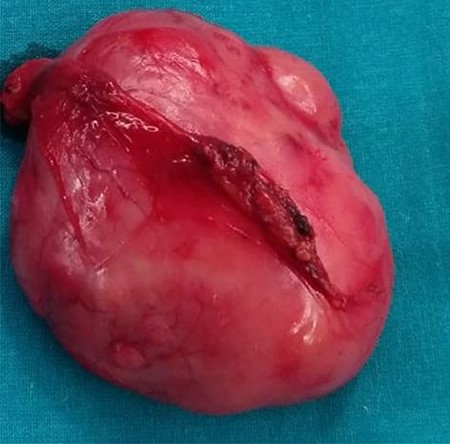
Macroscopic description: round and smooth brownish well delimited 6 cm in diameter solid lesion with homogeneous fibrous appearance. No signs of macroscopic necrosis are evident.
The pathologist report identifies a well-defined lesion 6 cm in diameter. Microscopically, fusocellular proliferation can be seen with hemangiopericytoid growth pattern, swirling and with abundant ‘deer antler’ shaped vessels. Absent necrosis is observed, and there are three non-atypical mitosis figures/40 high-power fields (HPF). It shows positivity for CD34, CD99, BCL2 and betacatenin (cytoplasmic) and negativity for CKAE1/AE3, desmin smooth muscle actin and S100. With these findings, benign TFS was diagnosed (Fig. 4).
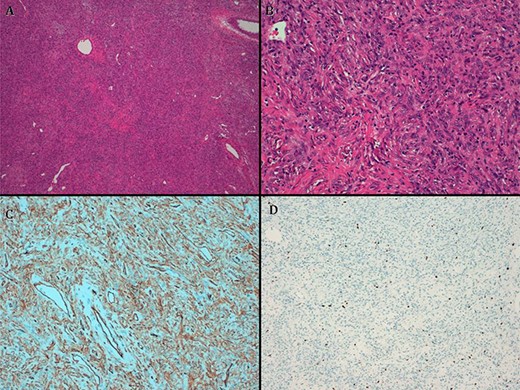
(A) hematoxylin–eosin, 4×. Slightly storiform diffuse proliferation. (B) HE, 20×. Fusiform cells with oval nuclei and extracellular collagen deposit. (C) CD34. (D). Ki67.
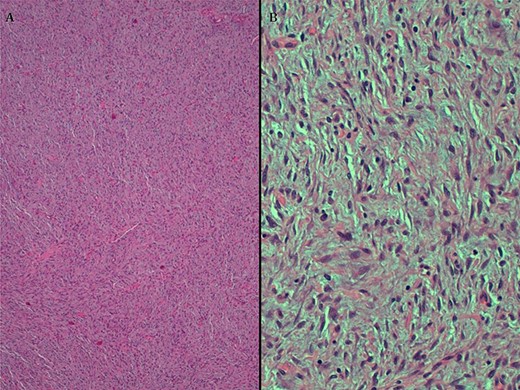
A. HE, 4x. Cells with diffuse growth. B. HE, 20x. Fusocellular neoplasia with clear eosinophilic cytoplasm and homogeneous elongated nuclei.
Free surgical margins were obtained as normal adipose tissue surrounding the tumor was shown.
After 2 years of follow-up, the patient had a favorable outcome without tumoral recurrence.
Case 2
A 21-year-old male patient came to the consult with a supraumbilical slow growing lump. The mass was well defined, tender, simulating a supraumbilical incarcerated hernia. It was scarcely painful at examination. No tenderness and no enlargement with Valsalva maneuver were present and digestive symptoms were absent. With the initial diagnosis of an incarcerated epigastric hernia versus abdominal wall tumor, a preferent open surgery was decided due to the low benefit of laparoscopy in this case.
During the procedure, a hard, well-defined nodular lesion of approximately 4 cm in diameter was found. Histological analysis (AP) described a benign mesenchymal tumor, suggesting a TFS with optimal surgical margins.
Microscopically monomorphic cell proliferation between lax fibrous stroma with abundant vascularization of small caliber, not encapsulated and with infiltrative edges was seen. Intertwined beams with fusiform and star cells, oval nuclei, fine chromatin and elongated eosinophilic cytoplasm were also present. Some collagen fibers between cells and Isolated non-atypical mitosis figures (0–1/10 HPF) were identified. Absence of necrosis. Immunohistochemically, positivity for CD34, CD99, bcl2 (cytoplasm) and smooth muscle actin were shown. Betacatenin, desmines and S100 were negative. Ki67 is 10–12% (Fig. 5).
Due to the results of FST, the case was discussed in the multidisciplinary tumors committee and follow-up as a low-risk tumor was made. After 1 year, the patient is asymptomatic and no recurrence has occurred.
DISCUSSION
TFS are rare mesenchymal neoplasms with a fibroblastic differentiation. Historically, they were described with different names (mesothelioma, pleural fibroma, submesothelial fibroma, subserosal fibroma and localized fibrous tumor). Hemangiopericytoma has been considered a different entity. However, the arrival of the IHQ diagnosis has led pathologists to consider it as the same entity, although the term of TFS is preferred [1].
Epidemiologically, about 2% of all soft tissue tumors are considered to be TFS [3]. The incidence is similar for men and women. Though they can debut at any age, they usually appear between the fifth and sixth decade of life.
They were first described by Klemperer and Rabin in 1931 at the thoracic region [4]. However, this tumor has been also registered in other body regions. TFS is generated in serous membranes and soft tissues. Approximately, 30% of cases are present in the thoracic cavity (including pleura, lungs and mediastinum). The peritoneal cavity, retroperitoneal soft tissues or pelvis represent the 30% of cases [5]. Approximately, 20% correspond to head and neck (including meninges). Less frequent locations are soft tissues of the trunk and extremities (10%) and bones.
The most frequent abdominal symptoms are palpable mass, weight loss and pain. Urinary or gastrointestinal symptoms have also been described due to compression. In the beginning, they are usually asymptomatic and sometimes when they are diagnosed, they have already reached a large size.
It is generally suspected after imaging tests. However, the final diagnosis requires histological confirmation.
The imaging tests required for diagnosis are CT scan or magnetic resonance imaging (MRI). Both show similar radiological characteristics to other soft tissue tumors, and there are no specific pathognomonic findings for TFS.
Its histological diagnosis is based on typically morphologic findings in conjunction with a characteristic immunophenotype. Complete resection of the tumor is necessary for proper study, and FNA is often not useful for diagnosis.
The purely histological differential diagnosis includes the deep fibromatosis (desmoid tumor), low-grade fibromyxoid sarcoma and myofibroblastic tumors. Desmoid tumors share many morphological features with TFS, but not its mutation of the APC gene that leads to overexpression of nuclear beta-catenin. Low-grade fibromyxoid sarcoma is another entity that should be ruled out when myxoid areas are observed, but in these tumors, the absence of MUC4 expression is characteristic. On the other hand, myofibroblastic tumors are usually positive for actin and/or desmines [6].
We consider essential that both the preoperative diagnosis and the postoperative evolution of the patient is to be made in a multidisciplinary way to achieve a correct scheduled treatment.
Size of TFS is quite variable. They are usually well defined, with a fibrous or serous pseudocapsule. Sometimes, they show multiple nodules and in general, excision of these tumors is easy.
Microscopically, they present fusiform cells, with elongated nucleus, dispersed chromatin and absent nucleolus, surrounded by scarce cytoplasm. The cells are surrounded by collagenous stroma. They frequently show a prominent vascularization, with hemagiopericytic growth pattern.
Immunohistochemistry has become the most useful tool to differentiate TFS from other tumors such as mesotheliomas or sarcomas [7]. Conventional markers usually include the expression of CD34, Bcl2, CD99, vimentin (in the absence of actin), desmine, S100 protein or epithelial markers.
Surgery in these tumors should be planned with the aim of achieving complete surgical resection with optimal margins as TFS can have a malignant behavior, and to avoid local recurrence.
The clinical behavior of these tumors is unpredictable. Most publications describe benign behavior, but between 15 and 20% of tumors produce metastases [8]. These patients have a worse prognosis with a 75% mortality at 5 years. Generally, meningeal TFS tumors have a more aggressive behavior.
Tumor recurrence is usually due to peritoneal, pleural or meningeal tumor seeding due to systemic or hematic extension, or incomplete resection. They usually metastasize to lung, liver, brain and bone.
Malignant criteria are considered for large tumors (110 cm), with hypercellularity, high mitotic activity (>4 mitosis per 10CGA), pleomorphism, presence of hemorrhage or necrosis, infiltrative borders or loss of CD34 [9].
The recommended treatment is tumor exeresis. Its management should be discussed in a multidisciplinary committee with experienced specialists in sarcomatous tumors. A preoperative vascular and arterial embolization study should be considered because of the risk of bleeding during excision [10].
Decision-making about the ideal surgical approach should be based on: experience of the surgical team, localization and size of the tumor. In our two cases presented in this manuscript, we decided an open approach based on the localization of both tumors.
There is no evidence that neoadjuvant or adjuvant radiotherapy is beneficial.
In particular, for patients with advanced disease (metastatic TFS or advanced local disease), treatment has not been well established.
It is very important to determine the malignant potential of the TFS and to establish an individualized follow-up. To determine the risk of malignancy of each tumor, a classification was performed by Demicco [11] in 2017, which classifies the TFS into three groups according to their risk of metastasis. According to the number of mitosis, age, size and presence of necrosis, the risk of metastasis is estimated, as is shown in Table 1.
Demicco’s malignancy risk classification
| N.° | Points | |
|---|---|---|
| Mitoses (per10 HPF) | 0 | 0 |
| 1–3 | 1 | |
| ≥4 | 2 | |
| Age | <55 | 0 |
| ≥55 | 1 | |
| Size | 0–4.9 cm | 0 |
| 5–9.9 cm | 1 | |
| 10–14.9 cm | 2 | |
| ≥15 cm | 3 | |
| Necrosis | <10% | 0 |
| ≥10% | 1 | |
| Risk sum stratification | Low | 0–3 |
| Intermediate | 4–5 | |
| High | 6–7 |
Depending on the malignant potential determined for each tumor, follow-up will be established based on guidelines for soft tissue sarcomas of the National Comprehensive Cancer Network: for low-risk tumors, an imaging test is recommended every 6 months for 3 years, and then annually until 5 years; for medium or high risk tests, every 3–4 months during the first 2 years and then every 6 months until the fifth year. After 5 years, more tests are not recommended since recurrence at that time is infrequent.
In conclusion, TFS are mesenchymal neoplasms more frequently in the pleural region, but can occur at any location. They are usually asymptomatic tumors. Diagnosis is led by imaging tests (CT scan and MRI) and surgical excision. Fine needle aspiration does not usually help the diagnosis. Surgical removal is the treatment of choice due to its malignant potential and is essential for proper diagnosis. Prior embolization can be considered in large and hypervascular tumors. The clinicopathological characteristics will allow us to classify its malignant potential and establish the follow-up scheme. It is important to suspect this type of tumors when well-defined and hypervascularized lesions are found. They should always be removed and followed closely according to their malignant potential.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}