





ABSTRACT
Malignancies characterized histologically by high-grade monotonous small round blue cells (SRBCs) belong to a heterogeneous group of neoplasms often referred to as Ewing family of tumors. The most common molecular confirmation of these neoplasms is by fusions between EWSR1 gene on chromosome 22 and the ETS family of transcription factors, including FLI1 gene (11q24) and the ERG (21q22), that are implicated in the development of different tissues as well as cancer progression. In this article, we present a case of highly aggressive extraskeletal SRBC tumor involving the foot of a 24-year-old male with sole molecular findings of mutations in KAT6A, NAV3 and SMARCA1 genes with high expression of soft tissue markers (COL1A1, COL1A2, COL3A1) and MYC mRNA. To our knowledge, this unique mutational pattern has not previously been described in SRBCs.
INTRODUCTION
Malignant neoplasms with high-grade monotonous small round cells, originally described in 1921 as ‘round cell sarcoma’ of unknown origin [1], occur mostly as primary tumors of long tubular bones in children and adolescents but may also be extraskeletal. Histologically similar neoplasms with divergent gene expression patterns are often referred to as Ewing-like family of tumors. These tumors are often associated with some characteristic immunohistochemical, genetic and molecular markers.
Chromosomal translocation-related genetic fusions between EWSR1 on chromosome 22 to the FLI1 gene on chromosome 11 and other genes encoding ETS family of transcription factor (EWS-ETS) are considered a genetic hallmark of human Ewing sarcoma [2–4]. Moreover, the epigenetic status of genes responsive to transcriptional regulation by EWS-ETS is important for Ewing sarcoma development and its phenotypic manifestation [5]. In a retrospective study of a large cohort of unclassified sarcomas with a round cell component, 10% were shown to harbor ‘CIC-DUX4’ or ‘BCOR-CCNB3’ fusions or other ‘CIC’ rearrangements [6]. Undifferentiated small cell sarcomas lacking these molecular and genetic markers, as in this case report, have also been referred to as ‘Ewing-like tumors’ [7].
CASE REPORT
A 27-year-old male presented with 3-month history of ‘callus’ in his left foot, which had increased in size over time, becoming more red, exquisitely tender and painful in the last few days. The lesion, 1.5 cm in diameter, involved the plantar surface of his forefoot where it was associated with pain described as sharp, increasing in intensity by pressure during walking. The pain decreased with rest but was tender, swollen, warm and red. The patient complained of fever and chills while at home.
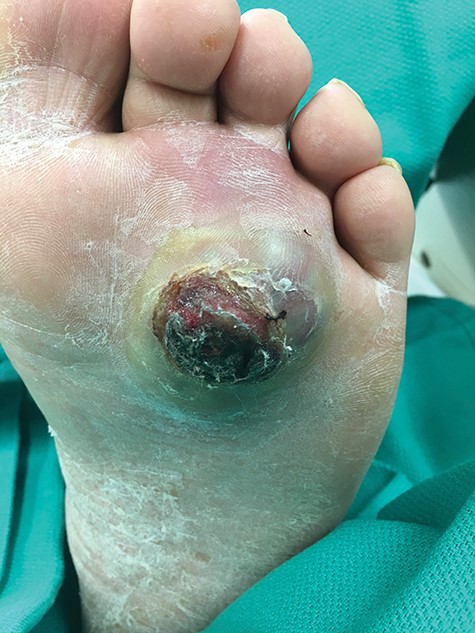
At presentation, the lesion appeared as a variably ‘hyperkeratotic mass with deep ulceration’.
On physical examination, the only significant findings included morbid obesity and a ‘small red tender lesion’ resembling an ‘abscess or fluid collection at the bottom of his foot’ (Fig. 1). X-ray of the patient’s foot was interpreted as ‘soft tissue swelling’ (Fig. 2). The patient was treated with antibiotics for the initial impression of cellulitis and sepsis.
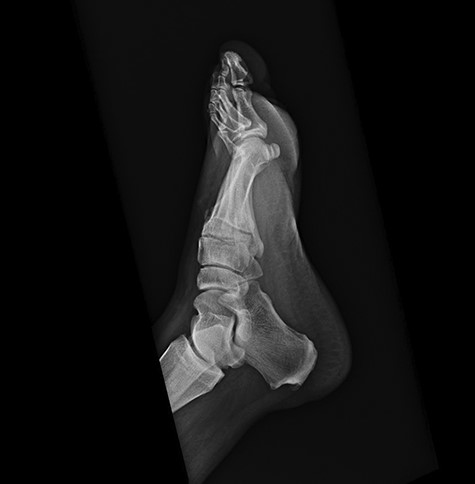
Initial x-ray of the foot was interpreted as ‘soft tissue swelling of the foot’.
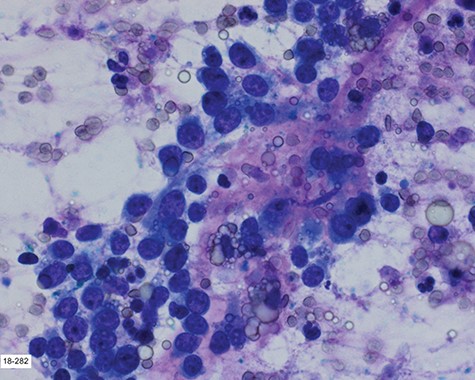
Touch prep of the excised lesion, prepared intraoperatively, showed loosely cohesive malignant neoplastic cells with small hyperchromatic nuclei and scant cytoplasm suspicious for a small round cell tumor.
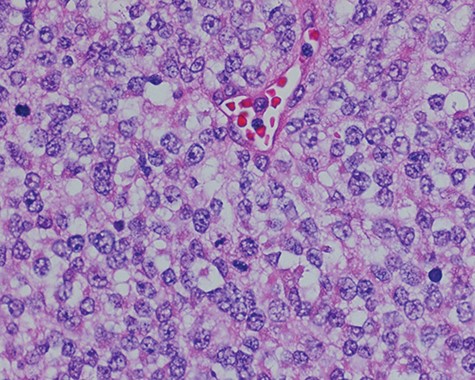
Sections of the lesion consist predominantly of sheets of poorly differentiated malignant neoplastic cells with moderately pleomorphic vesicular nuclei, clear cytoplasm, and abundant mitotic activity.
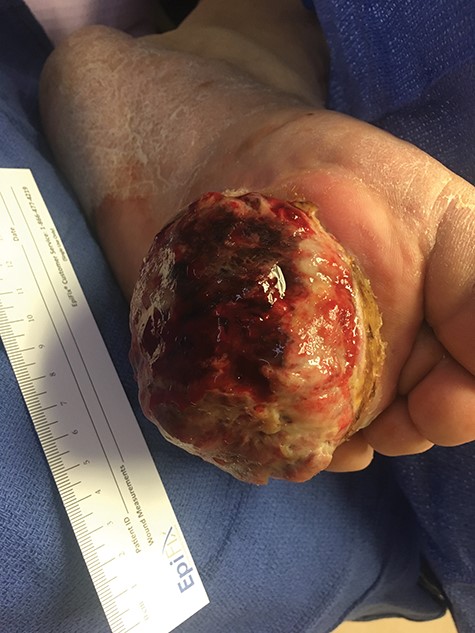
Two months following the excision, the patient presented with a significantly enlarged, necrotic and ulcerated tumor at the excision site.
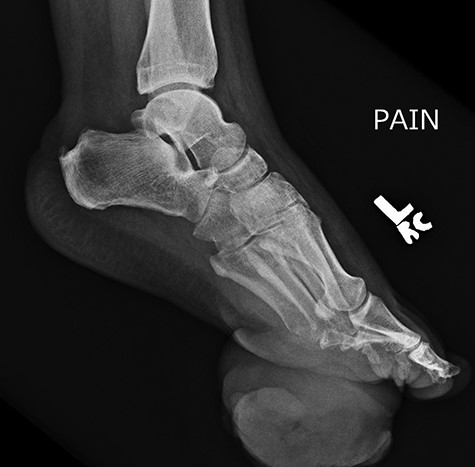
Repeat lateral X-ray views of the foot, 2 months later, showed ‘interval appearance of large soft tissue mass, plantar to the phalanges’. The differential diagnostic consideration by the imaging studies included ‘phlegmon, abscess, hematoma, or less likely a rapidly growing neoplasm’.
Surgical consultation revealed a deeply ulcerated cavitary-like mass with gelatinous content. The excised mass, covered by an ellipse of skin, was 5.0 cm × 3.5 cm × 3.5 cm. On section, the mass was 2.5 cm in diameter, pale tan-white with red–brown areas of discoloration. Intraoperative touch preparation of the lesional cut surface was suspicious for small round blue cell (SRBC) tumor (Fig. 3). Microscopic sections of the lesion consisted predominantly of sheets of poorly differentiated malignant neoplastic cells with moderately pleomorphic vesicular nuclei, scant clear cytoplasm and abundant mitotic activity (Fig. 4). The neoplasm extended to the inked surgical margins of resection.
Immunohistochemical stains of the lesion included the following results: (i) vimentin, CD 99, c-MYC and WT1 were diffusely positive; (ii) CD 56 was focally positive and (iii) CD10, CD45, S100, chromogranin, synaptophysin, CK-pan, HMB-45, myogenin, CD34, desmin, ERG and NKX2.2 were all negative.
Next-generation sequencing (NGS) testing of the tumor was negative for expression of fusion RNA or mutations involving EWSR1 gene. Subsequent comprehensive NGS molecular genetics testing of 1400 genes, were negative for any translocations that lead to the expression of fusion RNA, there was no evidence of a fusion RNA. Exons of 1385 cancer genes were sequenced at Neo-Genomics Laboratories (La Jolla, CA) and included 507 genes involved in fusions and more than 850 genes either mutated or deregulated in cancers. In addition, 160 bp of the 5′ and 3′ untranslated regions of every targeted gene were included in this testing.
As an overall summary of the available data and testing of this neoplasm, the following results were noted: (i) negative for specific chromosomal translocations; (ii) no evidence of a fusion mRNA involving CIC, Dux4, BCOR or any of the tested 1400 oncogenes; (iii) negative for SYT genes by fluorescence in situ hybridization, (iv) high expression of soft tissue markers (COL1A1, COL1A2, COL3A1) as well as MYC mRNA and (v) positive for mutations in KAT6A, NAV3 and SMARCA1 genes. Details of the KAT6A, NAV3 and SMARCA1 are as follows:

Approximately 2 months following surgical excision of the tumor, the patient presented with a significantly enlarged and painful recurrence of the mass (Fig. 5). Repeat X-rays of the foot showed ‘interval large soft tissue mass’ (Fig. 6). Nuclear three-phase bone scan was ‘suspicious for osteomyelitis or tumor involvement of bone’.
As a follow-up treatment, the patient underwent an above the knee amputation of left lower extremity and was subsequently found to have lung metastases within 1 year of initial diagnosis.
DISCUSSION
In this case of an undifferentiated SRBC tumor, the only mutations identified in the tumor were frame shift variants of SMARCA1, KAT6A and NAV3. All other tests including testing for a fusion RNA, 1400 oncogenes with an NGS Ewing sarcoma fusion profile, were negative. Although aberrant expressions of these frame shift variants have been described in a variety of tumors [8–10], they have not, to our knowledge, been previously described as the sole molecular/genetic findings in an undifferentiated SRBC tumor, especially involving the foot. The rapidly progressive course of this case, possibly partially or totally attributed to the high expression of soft tissue markers (COL1A1, COL1A2, COL3A1) as well as MYC mRNA, is indicative of the very aggressive nature of this tumor. Additional testing with possible retrospective molecular and mutational studies of such SRBC tumors are needed for clarification and understanding of their varied pathogenesis and classification.
Conflict of interest
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}