





Abstract
Solitary fibrous tumors (SFT) are uncommon fibroblastic mesenchymal neoplasms that display a wide range of histologic behaviors. These tumors, which are estimated to account for 2% of all soft tissue neoplasms, typically follow a benign clinical course. However, it is estimated that 10–30% of SFTs are malignant and demonstrate aggressive behavior with local recurrence and metastasis up to several years after surgical resection. We report a case of SFT arising from the stomach, which is an exceptionally rare finding and has been reported only six times in the literature. Our case was complicated by diagnostic dilemma with GIST, highlighting the challenges of diagnosing and characterizing SFTs. Additionally, this tumor was associated with dedifferentiation into undifferentiated pleomorphic sarcoma. To our knowledge, there are no documented cases of a malignant SFT arising from the stomach to demonstrate dedifferentiation into an undifferentiated pleomorphic sarcoma.
INTRODUCTION
Solitary fibrous tumors (SFT) are uncommon fibroblastic mesenchymal neoplasms estimated to account for <2% of all soft tissue neoplasms and occur typically in the fifth or sixth decade (1). These tumors usually follow a benign clinical course; however, it is estimated that 20% of SFTs are malignant and demonstrate aggressive behavior with local recurrence and metastasis up to several years after surgical resection (2). In this report, we present a rare case of a SFT of the stomach which was complicated by diagnostic dilemma with gastrointestinal stromal tumor (GIST), highlighting the necessity of including this tumor in the differential for neoplasms of the stomach. There were only six cases reported in the literature with none having features of dedifferentiation and early metastatic spread to liver (3).
CASE REPORT
A 68-year-old male presented to the emergency department with vague complaints of right-sided flank pain. The patient had a history of nephrolithiasis and underwent a CT abdomen (Fig. 1A). The scan revealed a large heterogeneous mass in the left upper quadrant. Subsequently, an MRI was performed, confirming a 16 × 9 cm2 complex mass with both cystic and solid components (Fig. 1B). This appeared to extend from the gastric wall and resulted in mass effect on the pancreas; however, there was no evidence of lymphadenopathy or metastatic disease. While this lesion was difficult to definitively characterize by imaging studies, a diagnosis of mesenchymal origin tumor such as a GIST was initially preferred.
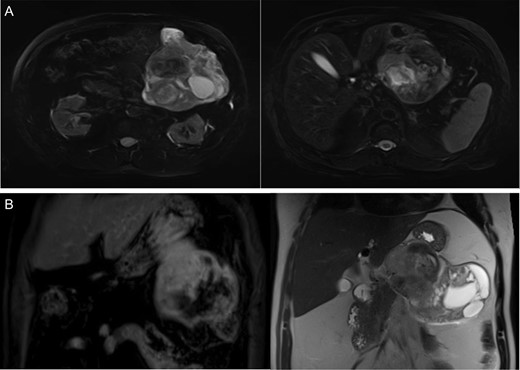
(A) CT abdomen demonstrates large heterogenous mass. (B) MRI coronal images display a heterogeneous mass on T1 (left) and T2 signal (right) arising from the submucosa with components restricting diffusion.
The patient underwent endoscopic ultrasonography with fine needle aspiration of the mass. The lesion appeared to arise from the submucosa, as the mucosa was intact overlying the mass. On cytology, a paucity of bland spindle cells was present (Fig. 2A) and the cell block contained several small clusters of cells which stained strongly for CD34, but failed to stain for DOG1 or CD117 (Fig. 2B). Given the small amount of material present, additional testing for PDGFR and succinate dehydrogenase testing was not able to be performed at this time to confirm the diagnosis of GIST. GIST remained the favored diagnosis given the morphology and location of the tumor. The options were to repeat the biopsy or start imatinib therapy with a plan of repeat imaging after short-term follow-up. Because of extensive necrosis of the tumor, repeat biopsy was felt to be a less favorable option and the patient chose to undergo treatment with short-term imaging follow-up. Accordingly, the patient began treatment with imatinib. However, after 4 weeks of therapy, there was no significant radiologic regression. A second biopsy was performed and the specimen was sent for STAT6 immunohistochemistry (Fig. 2C). This revealed diffuse strong nuclear positivity and a diagnosis of SFT was made.
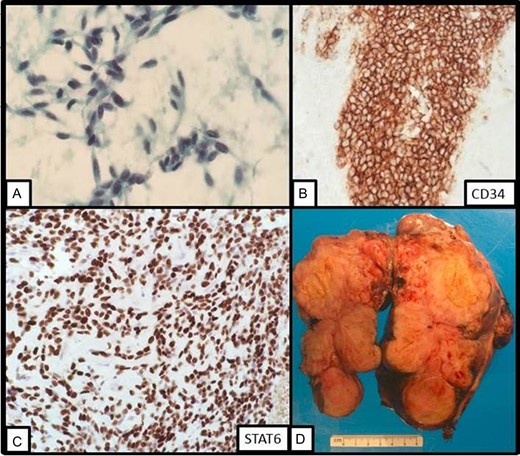
(A) On fine needle aspiration cytology, bland spindle cells are present. (B) On cell block, the lesional cells stains strongly with CD34 immunostain. (C) On repeat fine needle aspiration cytology, STAT6 displays strong nuclear staining. (D) Gross image of the resection specimen revealing a tan solid and cystic lobulated submucosal mass.
Patient underwent en bloc resection of the tumor along with partial gastrectomy involving the lesser curvature of the stomach. The mass was found to arise from the lesser curvature of the stomach with extension into the lesser sac (Fig. 2D). The patient’s postoperative convalescence was uncomplicated, aside from a mild self-limiting ileus. Histopathologic analysis revealed a predominantly spindle cell tumor with marked variability in cellularity with areas resembling fibrosarcomatous transformation. Furthermore, zones of frank necrosis, invasion into muscularis propria and regions of marked pleomorphism were consistent with an undifferentiated pleomorphic sarcoma. The tumor displayed brisk mitotic activity with up to 15 mitotic figures per 10 high power fields and the resultant French Federation of Cancer Centers Sarcoma Group (FNCLCC) was histologic grade 3/3. Notably, the resection margins were negative for malignancy. The patient was prescribed to undergo surveillance imaging in accordance with the National Comprehensive Cancer Network (NCCN) guidelines every 3–6 months for the first 2 years after surgery. The follow-up CT scan performed 4 months after surgery showed a new 2.3 cm hepatic lesion suspicious for metastasis; subsequent biopsy confirmed the diagnosis of metastatic disease. He later underwent left lateral segment resection with uneventful postoperative recovery. The pathology report confirmed metastatic high-grade sarcoma with margins negative for malignancy (Fig. 3).
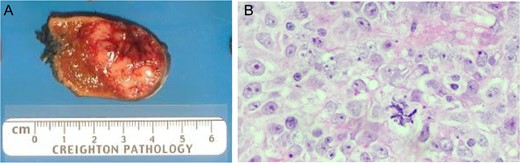
(A) Liver metastastectomy demonstrating ‘fish fleshy’ consistency of metastatic sarcoma. (B) Metastasic lesion demonstrating dense solid sheets of spindled to round high-grade pleomorphic nuclei with large, prominent nucleoli, numerous mitotic figures (including atypical, bizarre forms) and large areas of geographic necrosis.
DISCUSSION
SFT are rare mesenchymal tumors that typically follow a benign, indolent course with low incidence of metastasis (4). The immunophenotype of SFTs consists of strong and diffuse CD34 positivity, however, this is not specific as many other tumors stain for CD34, notably GIST (4). This feature along with the location and necrotic features made our differential diagnosis difficult. CD34 staining in SFT, however, may be patchy and sometimes absent, especially in cases of malignant SFT. Strong and diffuse nuclear staining for STAT6 is seen in most SFT, as a recent case series by Thway demonstrated strong nuclear STAT6 positivity in 206 out of 240 SFT cases examined. Nuclear staining of STAT6 was a specific finding associated with SFT, as both nuclear and cytoplasmic STAT6 activity was observed in other mesenchymal neoplasms (4).
Currently, the most favorable treatment option is complete surgical resection. The role of non-surgical options such as chemotherapy and radiation particularly in recurrent, metastatic or unresectable tumors are limited. Due to the rarity of the presence of dedifferentiation in SFT, there is insufficient data to accurately predict prognostic outcomes; however, few cases suggest the presence of a dedifferentiated component in SFT is associated with poor prognosis and suggests more aggressive clinical behavior than a malignant SFT without dedifferentiation (4).
Our case report highlights the challenges of diagnosing and characterizing SFTs. While our patient was treated with imatinib for probable GIST for 4 weeks before reanalysis with an additional biopsy, the steps made to correctly diagnose this patient on follow-up were imperative and highlight the importance of clinical judgment. STAT6 by immunohistochemistry is very specific for SFT and may have established this diagnosis earlier. Therefore, it is imperative to keep SFT, albeit exceedingly rare, in the differential diagnosis of mesenchymal neoplasms of the stomach.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}