





Abstract
Cystic teratomas are rare pluripotent embryonic tumors which most commonly originate in gonadal organs. Extra-gonadal cystic teratomas are exceedingly uncommon, accounting for only 1% of all cystic teratomas, and have been reported in unusual locations including the kidney, mediastinum and liver. These extra-ovarian cystic teratomas have also been known to harbor other neoplasms including carcinoid tumors. In this report, we describe a unique case of a hepatic cystic teratoma occurring as a combined tumor with a carcinoid in a young female. The patient underwent elective laparoscopic resection of her tumor after extensive radiographic and endoscopic work-up for chronic, non-localizable abdominal pain. We believe the carcinoid tumor arose de novofrom committed differentiation of a cell line within the teratoma, and not metastatic spread.
INTRODUCTION
Primary liver tumors are rare causes of chronic abdominal pain in the female population. A 1975 survey by the American College of Surgeons identified only 378 female patients in 477 hospitals over a 5-year period from 1970 to 1975; the majority having benign adenomas and focal nodular hyperplasia [1, 2]. Rarer still, are cystic teratomas of the liver; which account for <1% of all cystic teratomas reported in the literature [1, 2].
To the best of our knowledge, six case reports of hepatic cystic teratomas have been described in adults, mostly in female patients [3]. Hepatic cystic teratomas have a predilection to the right lobe and become clinically apparent due to the development of capsular pain from their mass effect [3]. In this case report, we describe a young female who presented with chronic abdominal pain and, after extensive work up, was found to have a hepatic cystic teratoma harboring a carcinoid tumor in segments 7 and 8 of the liver.
CASE REPORT
A 33-year-old female was referred for surgical evaluation for acute on chronic non-localizable abdominal pain. She was 1 month out from her sixth repeat caesarian delivery and her review of systems was otherwise negative. Her surgical history included a bilateral salpingo-oophrectomy done during the sixth caesarian delivery. The rest of her medical history was unremarkable.
Initial radiographic evaluation with computed tomography (CT) demonstrated a mass in the dome of the liver (Fig. 1a and b). A multiphase magnetic resonance (MR) scan of the abdomen followed and a 3.7 × 5 × 2 cm3 complex lesion was found, containing: cystic elements, macroscopic fat and a calcification (Fig. 1c and d). Percutaneous CT-guided biopsy was then performed and pathology showed atypical columnar epithelium with neuroendocrine differentiation. Both an upper and lower endoscopy did not identify any primary tumor. A positron emission tomography scan did not show evidence of metastatic disease or evidence of other pathology.
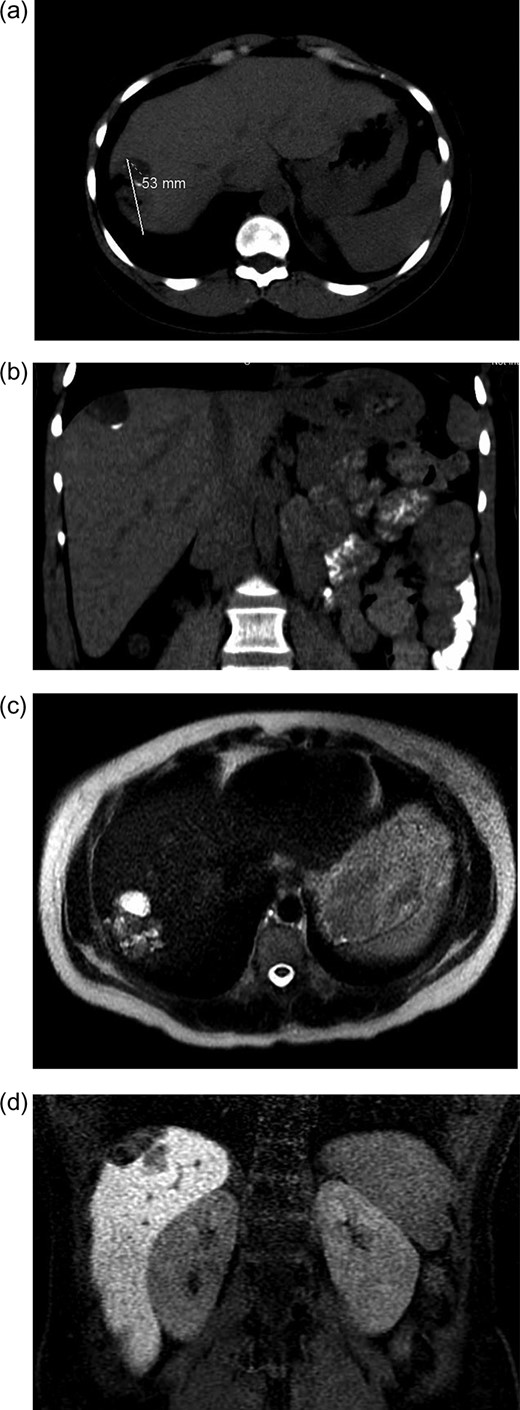
(a and b) CT abdomen and pelvis without IV contrast demonstrating tumor in axial (a) and coronal views (b). (c and b). MR abdomen and pelvis with and without IV contrast (Eovist, gadoxetate disodium, Bayer Pharmaceuticals) showing 3.7 × 5 × 2 cm3 complex lesion containing cystic elements, macroscopic fat and calcification in axial (c) and coronal (d) views.
Elective resection of her liver tumor was undertaken with partial hepatectomy 4 months after the initial CT scan. Resection was achieved laparoscopically.
Laparoscopic review of the abdomen did not reveal evidence of extra-hepatic disease. Grossly, the tumor appeared to have complex cystic and solid components involving the dome of the liver abutting the central tendon of the diaphragm. Intra-operative ultrasound confirmed that the tumor involved segments 7 and 8 of the liver. Resection of the tumor was then completed with a Habib bipolar laparoscopic resection probe (AngioDynamics Inc., Latham, NY, USA) and the LigaSure vessel sealing system (Medtronic Inc., Fridley, MN, USA). The tumor was 6 cm in transverse dimension and 3 cm deep and was delivered in toto by enlarging the umbilical incision. Grossly and microscopically, all tumor margins were negative. Hemostasis of the liver bed was achieved by applying FLOSEAL hemostatic matrix (Baxter International Inc., Deerfield, IL, USA).
DISCUSSION
Cystic teratomas are rare pluripotent embryonic tumors containing all three germ cell layers: endoderm, mesoderm and ectoderm [4]. Most commonly, they are found in the ovary, but other extra-gonadal locations have been reported, including the mediastinum, retroperitoneum, sacrococygeal region and liver [4–6]. One mechanism by which cystic teratomas develop in extra-gonadal sites is thought to be due to arrest in germ cell migration along the urogenital ridge during fetal development, although it has also been proposed that re-implantation of an ovarian teratoma into an extra-gonadal site can also occur [3].
Extra-ovarian cystic teratomas are known to harbor other neoplasms. In 1992, a hepatic cystic teratoma in a child was reported to harbor a hepatoblastoma [7]. To the best of our knowledge, there has been no other tumor co-occurrence involving a hepatic cystic teratoma [7]. Although malignant transformation within cystic teratomas most commonly involves squamous cell carcinoma (80%) and adenocarcinoma (6.8%), carcinoid tumors have also been reported [8].
Carcinoids are tumors of neuroendocrine origin which most commonly involve the respiratory (23%) and gastrointestinal tracts (73%) [8]. Other locations accounting for carcinoids in descending order of frequency include: ovary, uterus, testis, liver, gallbladder, breast, urethra, prostate and kidney [8, 9]. Carcinoid tumors have co-occurred with cystic teratomas of the kidney, ovary and testis [8, 10, 11].
The patient’s surgical pathology was consistent with a low-grade neuroendocrine neoplasm arising within a mature cystic teratoma. The neoplasm took up 25% of the tumor volume and the rest was occupied by elements of a mature cystic teratoma. Histologically, squamous epithelium, sebaceous glands, thyroid, salivary gland, urothelium and gastrointestinal epithelium were all demonstrated (Figs 2–4). Final immunohistochemical analysis showed that the neuroendocrine neoplasm was reactive for: synaptophysin, AE1/AE3, STAT B2 and non-reactive for: CK 20, GATA3, CDX-2, CK 7, PAX 8, TTF 1, p 16, mammoglobolin, glypican 3, beta catenin, CD 117, DOG 1 and inhibin; thus, supporting its neuroendocrine origin from gastrointestinal tissue and consistent with a carcinoid tumor [12].
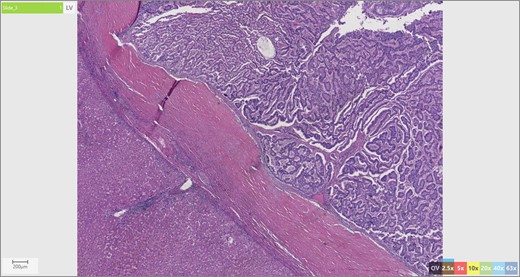
Liver parenchyma abutting neuroendocrine tumor.
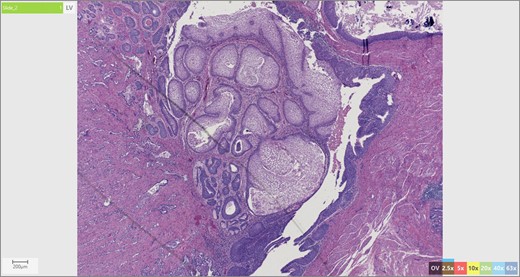
Skin adnexae and glandular elements of the mature cystic teratoma.
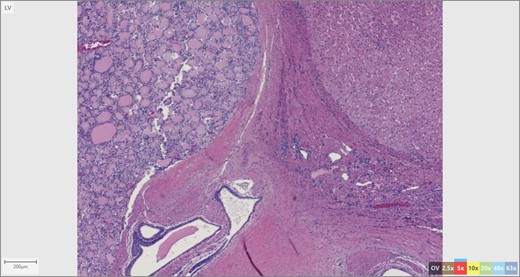
Thyroid tissue (left) adjacent to liver parenchyma.
We contend that the carcinoid tumor most likely arose de novo from committed differentiation of gastrointestinal tissue within the teratoma. Our exhaustive work up involving upper and lower endoscopy, cross-sectional imaging as well as the tumor immunohistochemistry makes the possibility of distant metastatic spread from another site highly unlikely. This is relevant in terms of management and prognosis; we believe that the tumor arose primarily and the resection to be curative.
Acknowledgements
I would like to thank Dr Jamie Donnelly for help in preparing the pathologic slides and Dr Jonathan M. Saxe for review of the article.
Conflict of Interest statement
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}