





Abstract
Paratesticular leiomyosarcoma is a rare tumour. It is seldom diagnosed pre-operatively and subsequent secondary resection is often required. Current treatment consensus comprises inguinal radical orchidectomy with high ligation of the spermatic cord. We present a case of a 74-year-old male with a 3-year history of a painless right-sided scrotal mass which following excision was found to be an epididymal leiomyosarcoma. A review of literature and treatment is presented in this article.
INTRODUCTION
Paratesticular sarcomas are rare, with only 110 cases reported in literature [1]. The most common histological subtypes include liposarcoma, leiomyosarcoma (LMS) and rhabdomyosarcoma. LMS is thought to arise from paratesticular smooth muscle tissues and may invade locally through direct tissue infiltration, or spread to distant sites via haematogenous or lymphatic routes. No established treatment protocol exists, however, general consensus comprises inguinal radical orchidectomy and high cord ligation, with some case series advocating adjuvant radiotherapy.
CASE REPORT
A 74-year-old male with a background of ankylosing spondylitis and hypertension presented with a long-standing, painful right-sided scrotal mass that had noticeably increased in size in the preceding 6 months.
He had undergone ultrasound investigation for the same complaint 3 years prior to his referral which demonstrated a bulky right epididymis with focally increased vascularity, thought to be a benign sperm granuloma (Fig. 1). Repeat USS 3 months later again showed some inflammatory thickening of the right epididymis and no further action was taken.
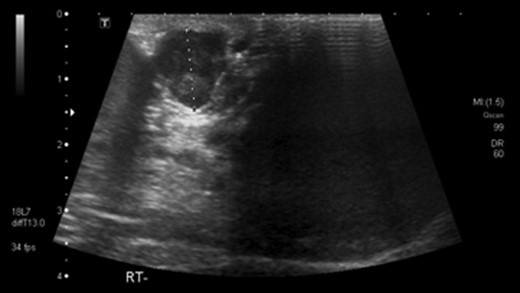
Ultrasound scan showing paratesticular leoimyosarcoma 3 years prior to referral.
At the time of referral, the lesion had dramatically increased in size and become more symptomatic, therefore surgical excision was agreed. He underwent a trans-scrotal right epididymectomy where an irregular white mass measuring 15 × 46 × 35 mm3 was excised. Histological examination demonstrated a well delineated mass comprising fascicles of cytologically malignant spindle cells (Fig. 2). These showed moderate to nuclear pleomorphism, frequent mitotic figures, including atypical forms, and zones of coagulative necrosis (Fig. 3). Immunohistochemistry showed the tumour cells were strongly and diffusely positive for desmin, smooth muscle actin and h-caldesmon. FISH analysis for MDM2 amplification excluded a dedifferentiated liposarcoma, the main differential diagnosis at this site. Overall, the features were those of a paratesticular LMS, which was FNCLCC/Trojani grade 2.
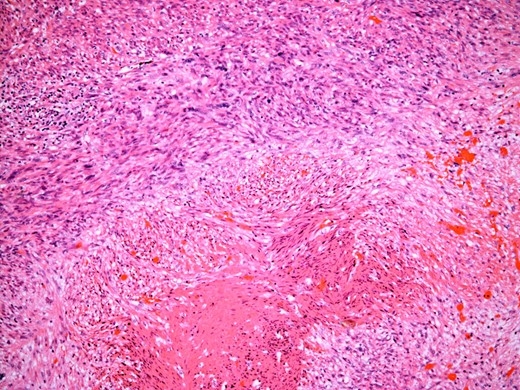
The tumour comprises fascicles of atypical spindle cells showing prominent cytological atypia and nuclear pleomorphism. Zones of coagulative tumour cell necrosis are present.
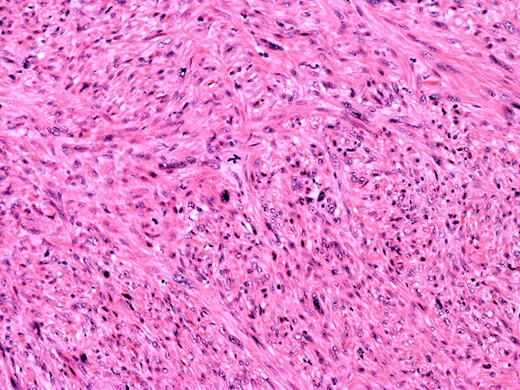
On higher power, severe cytological atypia is evident, with frequent mitotic figures including atypical mitotic forms.
Following a sarcoma MDT meeting, he was subsequently referred to a tertiary cancer centre for further management, undergoing a right inguinal radical orchidectomy with high ligation of the spermatic cord and wide excision of right-sided scrotal tissue and skin. Further histological analysis demonstrated no evidence of residual sarcoma in the final resection specimen.
DISCUSSION
Sarcomas of the genitourinary tract are uncommon and represent only 1–2% of all urological malignancies [1]. Less common are sarcomas of the paratesticular region, comprising tissues such as the epididymis, spermatic cord, inguinal canal and testicular tunica [1]. Localization to the epididymis is extremely rare (accounting for 4% of all paratesticular sarcomas). Paratesticular LMS is thought to arise from smooth muscle structures such as the wall of the epididymis or vas deferens, the cremaster muscle or the contractile tissues of the tunica. As paratesticular sarcomas are uncommon, limited data exists on the natural history of the disease or long term results of treatment.
The first case of paratesticular sarcoma was reported in 1845 by Lesauvage [2]. The most common histological subtypes are liposarcoma (20–32%), LMS (19–32%) and rhabdomyosarcoma (11–24%) [3]. Only 110 cases of LMS confined to the spermatic cord have ever been reported [1].
LMS has a peak incidence in the sixth and seventh decade of life [4], and usually presents as a slow-growing discrete mass entirely separate from the testis, which may or may not be painful and can be associated with a hydrocele [4].
Investigation should begin with ultrasound scanning (USS), which is the primary choice of imaging for any scrotal abnormality. USS has a sensitivity of 95–100% for distinguishing between intra- and extra-testicular lesions [5]. However, the distinction between benign and malignant paratesticular tumour is rarely made pre-operatively.
Due to the rarity of paratesticular LMS, no treatment protocol exists. Current consensus comprises radical orchidectomy with high ligation of the spermatic cord [5]. Definite diagnosis requires histological examination. LMS is assessed and graded according to mitotic rate, the percentage of necrosis and degree of nuclear pleomorphism.
As sarcomas of all grades have a tendency to infiltrate local tissues, adequate initial surgical resection can be difficult and regional recurrence is a major problem, with scrotal recurrence rates as high as 25–37% [6]. Adjuvant radiotherapy has been shown in one case series to reduce these rates of recurrence. Fagundes et al. [7] found recurrence in five of nine patients treated with orchidectomy alone and no recurrence in nine patients who received adjuvant radiotherapy. These findings are consistent of Catton et al. [8] who also found reduced loco-regional recurrence after adjuvant radiotherapy. Given these high rates of recurrence, long term follow up of paratesticular sarcomas following radical orchidectomy is paramount.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}