





Abstract
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome leading to colorectal cancer. This disease appears as a result of germline mutation in adenomatous polyposis coli (APC) gene. The aim of the present study is to report the association between two different nucleotide substitutions detected in a family with FAP. In the proband, p.His1172Gln (c.3516delT) was detected in exon 15 of the APC gene. Furthermore, p.His1172Gln (c.3516delT) and, in addition to this mutation, p.Met1413Val (c.4237 A > G) were detected in exon 15 in both daughters of the proband. However, we believe that single nucleotide change in codon 1413 may be a polymorphic variant and deletion T in codon 1172 of APC gene is associated with FAP, attenuated FAP and extracolonic FAP involvement. Along with common use of genetic tests in the clinical practice, genotype–phenotype correlation may be recognized better and useful for early diagnosis and prevention of familial cancer syndromes.
INTRODUCTION
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome, which is diagnosed by detection of adenomatous polyps over 100 leading up to colon cancer. Colorectal carcinoma and different extra colonic involvements are observed in the patients with FAP. Hundreds and even thousands of polyps appear in the colorectal area during second and third decades in the patients with FAP. These polyps transform into colorectal carcinoma around 40–50 years of age. Approximately 10% of the patients with FAP have polyps <100, and these transform into carcinoma during elder ages. This disease group is called attenuated FAP (AFAP).
FAP appears as a result of germline mutation in adenomatous polyposis coli (APC) gene [1]. APC is a tumor suppressor gene. About 60–70% of APC mutations are micro-deletions, and 10–15% are large deletion [2]. Fresh mutation incidence rate is higher in the patients with FAP. De novo mutation is observed in 25% of all patients [3]. More than 3000 mutations were defined in the APC gene [4]. Genotype–phenotype correlations of the patients with FAP could not be clearly determined. Individuals with same germline APC mutation may present different clinical findings due to the presumably environmental or other genetic factors [5, 6]. We reported two different nucleotide substitutions of APC gene detected in the patients with FAP phenotype in the same family.
CASE REPORT
The proband was a 54-year-old male patient who was referred to Department of General Surgery because of progressing rectal bleeding, weight loss, diarrhea and constipation. An ulcero-vegetating mass was palpated on 2.5 cm distant to the anal verge by rectal palpation. Polypoid lesions over 100 diffused along whole colon from terminal ileum were observed in the colonoscopy. Adenocarcinoma developed on FAP was detected in pathological examination of the polypectomy samples. Total proctocolectomy and terminal ileostomy were performed. Low-grade dysplasia and tubulovillous adenoma were detected in the periampullary region in the pathological examination of stomach and duodenum samples obtained during endoscopic examination of the upper gastrointestinal tract. There was not any other extracolonic organ involvement detected in the patient. The patient has two daughters who are 24 and 18 years of age, respectively. Both daughters had colonoscopy. During the colonoscopy, more than 100 polyps diffused along the colon were observed in the elder daughter, whereas more than 10 polyps were observed in the younger daughter. Low-grade dysplasia and tubular adenoma were detected in both daughters in the pathological examination of the polyps. The FAP patient and his family were referred to Department of Medical Genetics for genetic analysis. A detailed family history was obtained from the FAP family, and the pedigree of proband's family was drawn (Fig. 1). (We also wanted to examine proband's brother who is 51 years old. Unfortunately, he refused medical examination and genetic analysis.) Informed consent was obtained from all the family members included in the study. APC gene sequencing analysis was performed. In the proband, p.His1172Gln (c.3516delT) was detected in exon 15 of the APC gene (Fig. 2). Mutations of p.His1172Gln (c.3516delT) and p.Met1413Val (c.4237 A > G) change in exon 15 were detected in both daughters. APC gene mutation analysis was done to the mother to reveal whether p.Met1413Val (c.4237 A > G) change detected in the daughters is associated with clinical presentation of FAP. Same nucleotide change with the daughters, p.Met1413Val (c.4237 A > G), was detected in the mother. No pathological finding was detected in the colonoscopy of the mother.
![Pedigree of FAP family. Affected family members are identified by filled symbols; III-8 proband [p.His1172Gln (c.3516delT)], III-9 p.Met1413Val (c.4237 A > G), IV-3. The daughter with FAP [p.His1172Gln (c.3516delT) and p.Met1413Val (c.4237 A > G)]; IV-4 the daughter with AFAP [p.His1172Gln (c.3516delT) and p.Met1413Val (c.4237 A > G)].](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/jscr/2015/9/10.1093_jscr_rjv118/2/m_rjv11801.jpeg?Expires=1788215559&Signature=KaK6HGh5tC7OKG7M6gdlwMcpIQJuOhPkcybrErsn-UQ~BDau4R6RrL-RE0G0V860yuH0ESWt0rvm~XF9iyBDs2bAokbmdz4-5l5o8tjFsNY4YQ4vOcsQd-C4YHz3inp6qhPGf7XxmgtRwjvr-Prgs624ME2SkR9d5tFXXI2ocntgQbM4YoAtpnHmu3x5eRCkn3vWKIFErUMNmD1F-Jcg6tfPMZRHHW8IdCYvDjRw0Wz-gwNU0UEusrFXSfOX3b-RK84vCGOyWLgLaIybntenMwLtUiTfw1rZy1rGvn2UAWo-8RsAIejjmDQgYb0e3If9goVMmssVYme5U74DiLgD7A__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Pedigree of FAP family. Affected family members are identified by filled symbols; III-8 proband [p.His1172Gln (c.3516delT)], III-9 p.Met1413Val (c.4237 A > G), IV-3. The daughter with FAP [p.His1172Gln (c.3516delT) and p.Met1413Val (c.4237 A > G)]; IV-4 the daughter with AFAP [p.His1172Gln (c.3516delT) and p.Met1413Val (c.4237 A > G)].
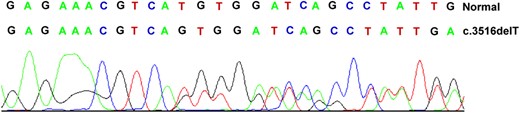
Normal and mutant sequences of the APC gene mutation in codon 1172.
DISCUSSION
FAP is an autosomal dominant disease, which is predisposing for colorectal cancer. Mutations in APC gene cause both classic FAP and AFAP. Sometimes, AFAP may imitate classic FAP. In such case, mutation analysis is useful. Because some mutations in the APC gene are associated with genetic heterogeneity, the type and severity of disease. However, APC mutation is not detected in 30–50% of the patients with FAP or AFAP phenotype [7]. Wide genomic deletions are observed in 10–15% of the patients with negative classic FAP mutations. These deletions are not found in the patients with AFAP [2]. In the present study, same APC deletion was detected in the proband and his daughter who presents classic FAP phenotype as well as his daughter with AFAP phenotype. In the analysis conducted by Tunca et al. [8] on a Turkish patient with colorectal cancer, T deletion in codon 1172 of which we have found was detected. This is a pathological mutation located in exon 15. Deletion of nucleotide T in codon 1172 causes frameshift mutation and creates a premature stop codon. Same APC gene mutations may cause different phenotypes of diseases by effect of environment and other genetic factors. Similarly, the p.His1172Gln (c.3516delT) mutation found in our family caused both FAP and AFAP phenotypes as well as tubular adenoma on the periampullary region in the proband. Upper gastrointestinal polyps exist in many of the patients with FAP. These polyps were associated with mutations in different regions of the APC gene. A significant association was reported between codon 564–1465 mutations and duodenal polyp and gastric adenoma in particular [9]. The T deletion in codon 1172 in our proband caused FAP phenotype as well as adenoma in the periampullary region.
The nucleotide change in codon 1413 existing in the mother and both daughters was detected in patients with both non-FAP, non-MAP (MUTHY-associated polyposis) and in the healthy controls in the study conducted by Azzopardi et al. [10]. Since the mutation was detected in the symptomatic (in both daughters) and asymptomatic (the mother) individuals in the family, we believe that such nucleotide change is a polymorphic variant.
Consequently, the FAP disease has a high incidence for development of cancer. Along with common use of genetic tests in the clinical practice in the future, genotype–phenotype correlation may be recognized better and useful for early diagnosis and prevention of familial cancer syndromes.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}