





Abstract
Cronkhite-Canada syndrome is an extremely rare condition of gastrointestinal polyposis in which the main presenting features are diarrhoea and dysgeusia. The polyps in this condition are characteristically distributed throughout the entire gastrointestinal tract except the oesophagus, and these patients exhibit unique ectodermal abnormalities. Herein, we report a 50-year-old male who had recurrent episodes of severe haematochezia from the polyps in the colon. Further examination and investigations revealed a diagnosis of Cronkhite-Canada syndrome. Proctocolectomy was carried out for medically refractory haematochezia and the patient is asymptomatic at present.
INTRODUCTION
Haematochezia in an adult raises significant concern among patients and physicians. Common causes of haematochezia in adults are diverticular disease, vascular ectasia, colitis (ischaemic, inflammatory or infectious), colonic neoplasia and anorectal conditions (1). Haematochezia is not reported as a common mode of presentation among patients with Cronkhite-Canada syndrome (CCS) in which diarrhea and dysgeusia are the predominant initial presentations (2). Surgery in patients with CCS has been carried out for associated gastrointestinal tract malignancies, intestinal obstruction and severe protein loosing enteropathy (3).
We present a case of persistent severe haematochezia in a patient with CCS, successfully managed with Proctocolectomy.
CASE REPORT
A 50-year-old Sri Lankan man presented with a 3-month history of diarrhoea, haematochezia and weight loss of 20kg. One month prior to these symptoms he has noticed dark pigmentation of the skin and hair loss. Physical examination revealed a cachectic man with alopecia, generalized skin hyperpigmentation involving the palms and onychodystrophy of fingernails (Fig 1–4).
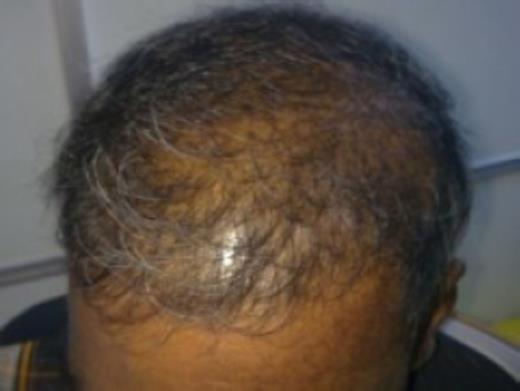
Alopecia
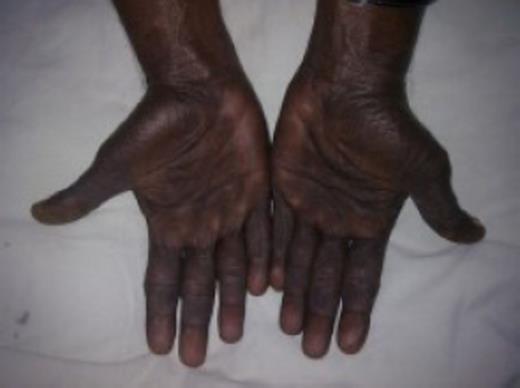
Diffuse skin hyperpigmentation involving the palms
Biochemical investigations revealed a haemoglobin value of 9g/dl and marked hypoalbuminaemia with serum albumin of 12g/l (normal- 36-44g/l). Upper gastrointestinal endoscopy and colonoscopy revealed multiple, sessile polyps of varying sizes distributed throughout the stomach, duodenum, colon and rectum without any polyps in the oesophagus. Video capsule endoscopy revealed numerous similar polyps throughout the small intestine (Fig 4).
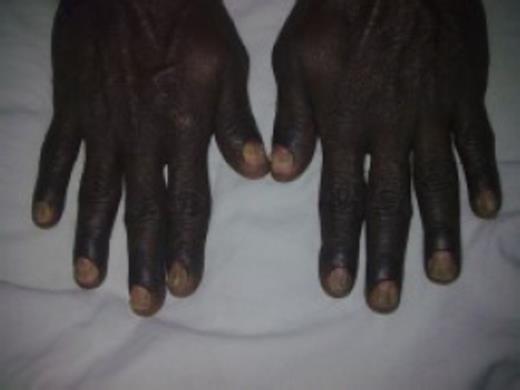
Onychodystrophy of finger nails
Biopsy of colonic polyps revealed cystic dilatation of crypts with oedema of the lamina propria suggesting hamartomatous polyps. Thus, a diagnosis of Cronkhite-Canada syndrome was made, considering the characteristic ectodermal abnormalities on physical examination, endoscopic findings and histology. The patient was commenced on oral Prednisolone 40mg daily and oral Zinc supplements. Haematochezia worsened despite medical treatment for 2 months and proctocolectomy with a permanent ileostomy was performed. Histology of the specimen revealed multiple polyps with a diverse spectrum of morphologic features including juvenile-type polyps, adenomatous polyps, serrated adenomas, hamartomatous polyps and mixed polyps.
DISCUSSION
First described in 1955, Cronkhite-Canada syndrome (CCS) is a rare, nonhereditary polyposis syndrome with an uncertain aetiopathogenesis. Approximately about 400 cases have been reported in the literature with the majority being reported from Japan. It is characterized by the presence of diffuse gastrointestinal polyposis (sparing the oesophagus) associated with characteristic ectodermal abnormalities such as alopecia, nail dystrophy and cutaneous hyperpigmentation (4,5).
The aetiology of CCS is currently unknown. Associations with raised ANA and IgG4 levels, hypothyroidism and various autoimmune diseases such as systemic lupus erythematous, rheumatoid arthritis and scleroderma have been published in the literature (6).
Most of these patients present with diarrhea and dysgeusia and the dermatological manifestations are known to appear later (2). The presentation in our patient is different because he had not developed dysgeusia, his cutaneous symptoms have preceded the onset of diarrhoea and haematochezia was one of his key presenting features.
There is no optimal treatment recommended for these patients. Up to now, different treatment modalities have been attempted with varying degrees of success (7). These include hyperalimentation, corticosteroids, H2-receptor antagonists, antibiotics, acid suppression, Cromolyn Sodium, anabolic steroids, surgery, and combinations of these therapies (7).
Among the various complications reported, the risk of developing gastrointestinal malignancies is significant. This can be as high as 15% and both gastric and colorectal malignancies are known to occur in these patients (8,9).
The long-term outcome of CCS is poor. Some studies have shown overall mortality rates as high as 55% among their patients (2).
To our knowledge, this is the first case of total proctocolectomy for refractory haematochezia in a patient with CCS. This also highlights the importance of considering Cronkhite-Canada syndrome as a rare possibility in a middle aged male presenting with haematochezia, specially if the patient is of Asian origin and if there are any cutaneous abnormalities suggestive of this condition.

{kind=link}
{kind=link}
{kind=link}