





Abstract
Thalassemia is one of the most prevalent monogenic disorders in low- and middle-income countries (LMICs). There are an estimated 270 million carriers of hemoglobinopathies (abnormal hemoglobins and/or thalassemia) worldwide, necessitating global methods and solutions for effective and optimal therapy. LMICs are disproportionately impacted by thalassemia, and due to disparities in genomics awareness and diagnostic resources, certain LMICs lag behind high-income countries (HICs). This spurred the establishment of the Global Globin Network (GGN) in 2015 at UNESCO, Paris, as a project-wide endeavor within the Human Variome Project (HVP). Primarily aimed at enhancing thalassemia clinical services, research, and genomic diagnostic capabilities with a focus on LMIC needs, GGN aims to foster data collection in a shared database by all affected nations, thus improving data sharing and thalassemia management. In this paper, we propose a minimum requirement for establishing a genomic database in thalassemia based on the HVP database guidelines. We suggest using an existing platform recommended by HVP, the Leiden Open Variation Database (LOVD) (https://www.lovd.nl/). Adoption of our proposed criteria will assist in improving or supplementing the existing databases, allowing for better-quality services for individuals with thalassemia.
Database URL: https://www.lovd.nl/
Introduction
Global epidemiology of hemoglobinopathies including thalassemia
There are approximately 270 million carriers of abnormal hemoglobins and thalassemia worldwide, with 80 million of them carrying β-thalassemia [1]. However, these numbers do not fully represent the overall impact of thalassemia, which not only affects individuals with symptoms but also their families, leading to socio-economic challenges.
The resistance of hemoglobinopathy carriers to malaria has caused an increase in alleles in historic malaria regions, including large parts of Africa for sickle cell disease (SCD) and for thalassemia a geographic belt stretching from sub-Saharan Africa and the Mediterranean into the Middle East and South-East Asia [2]. In LMICs located in this thalassemia belt, the pressure of malaria has resulted in thalassemia becoming the most prevalent genetic disease. While some LMICs have made progress in reducing thalassemia cases, the majority are still striving to control the disease and lessen its impact on health [3].
Thalassemia and its overriding challenge for low- and middle-income countries
Hemoglobinopathies are disorders that affect the expression of the α-globin (HBA) or β-globin (HBB) components of hemoglobin. SCD is the most common type caused by a variant of HBB that promotes the formation of hemoglobin aggregates at low oxygen pressure. β-thalassemia, on the other hand, is characterized by low production of β-globin. These disorders have a well-understood pathophysiology but present a growing global health challenge due to migration and new genotypes. SCD and thalassemia severity are determined by a combination of hundreds of different globin variants and genetic modifiers, which makes phenotype predictions difficult, even when HBA and HBB genotypes are known.
Individuals with thalassemia may have severe anemia, requiring costly blood transfusion and iron chelation for survival. Without treatment, severe cases can lead to death in the first decade of life. Adequate blood transfusion and monitoring, along with proper iron-chelation therapy, are crucial for good quality of life with minimal complications. However, providing safe blood and coordinating national-level transfusion and therapy is challenging in LMICs, where the majority of transfusion-dependent thalassemia (TDT) patients reside [4] and where approximately 80% of newborns with hemoglobinopathies are born [5, 6].
Thalassemia management involves an accurate diagnosis and comprehensive clinical care, including emotional, social, and financial support. Diagnosis requires a comprehensive clinical history, hematology and hemoglobin analyses, and molecular tests. However, access to diagnostic tests is sometimes limited, particularly in LMICs. What is more, with growing populations and improving healthcare facilities, the disease burden is expected to increase due tooverwhelming transfusion and budget capabilities for adequate thalassemia patient care, unless effective prevention programs are implemented.
Thalassemia management and control
HICs have successfully implemented national hemoglobinopathy control programs, but LMICs face financial, political, and infrastructural challenges in doing so [7]. Some LMICs, such as Turkey and Malaysia, have reported a reduction in thalassemia cases [8–10], while others, such as Pakistan and Iraq, continue to struggle despite implementing prevention programs. A systematic categorization of LMICs according to established thalassemia supports infrastructures [7] and the comparison of effective with ineffective control efforts may guide future programs. The failure of prevention programs in some countries is due to factors such as lack of training, limited awareness, poor coordination, and inexperienced support organizations [11].
Hemoglobinopathies are expected to become more prevalent due to poor management and migration of affected populations to non-endemic regions. This will result in an increasing health burden in HICs where hemoglobinopathies are frequently underdiagnosed, and health systems are often unresponsive. Poor coordination of international efforts and a lack of supportive infrastructures will exacerbate this issue, which may impose a strain on the health systems of both LMICs and HICs, increasing the global health burden of hemoglobinopathies [6].
Diagnostic laboratories in industrialized countries are categorized into tiers 1–4 based on the level of specialization, whereas LMICs aim to achieve an integrated network of tiered labs that provide broadly accessible, sustainable, and high-quality pathology services. Ethiopia’s model has found endorsement in the Freetown Declaration of October 2015 as a basis for effective healthcare [12]. Bilateral international support and geographical proximity can also improve healthcare in LMICs, as shown by Thailand’s support for Laos PDR and the establishment of thalassemia diagnosis in Laotian peoples [13].
Poor records on cases and trends
Importantly, the existence of a database of core information can guide prevention and management strategies for endemic genetic diseases. Countries on the thalassemia belt, including Bangladesh, Sri Lanka, Brunei, Laos, and Myanmar, have varying levels of success in disease control and report a lack of vital data on thalassemia carriers, affected births, and disease prevention initiatives [7, 14, 15]. In particular, little is known about hemoglobinopathy disease prevalence, clinical course, mortality, co-morbidities, and treatment outcomes in Bangladesh [15]. Sri Lanka has implemented a screening program to identify thalassemia carriers and increase awareness, but there is no evidence of its impact on reducing the number of births affected by thalassemia. The absence of a national registry or record of patient numbers and distributions may contribute to such lack of observable impact, hindering prevention efforts and effective resource management [14].
Tackling hemoglobinopathies
Economic burden of thalassemia
Economic burden-of-disease (BOD) or cost-of-illness (COI) studies help finance and plan a country’s health program [4, 16]. A COI study catalogues, quantifies, and calculates a problem’s combined costs to assess a disease’s economic impact on society [17]. Multiple countries have reported the economic burden of thalassemia. The anticipated annual cost for a thalassemia patient in Sri Lanka, India, Thailand, and Iran was reported as USD 2601 [18], USD 1135 [19], USD 950 [20, 21], and USD 833 [20], respectively, which is comparatively low compared to that for some other LMICs. However, in Malaysia, the estimated annual cost of α- and β-halassemia from treatment amounts to USD 10 499 [4]. Malaysia follows international thalassemia treatment guidelines similar to those in the UK and the USA[4]. Since most thalassemia patients receive regular blood transfusions and free iron-chelation therapy, Azman and colleagues (2016) found that Malaysia has a higher economic burden than other Southeast Asia (SEA) countries. By contrast, cost-effective screening and prevention programs have reduced thalassemia births in Cyprus, Israel, Sardinia, and Singapore [22–24] and make exceptional levels of care affordable. With their cost reduction compared to life-long therapy, comprehensive national screening programs are thus essential for economic growth [4].
The burden of thalassemia and hemoglobinopathy in India
β-thalassemia is the most prevalent hemoglobinopathy in India, which globally occupies a significant position in the thalassemia belt. India has the highest number of patients with thalassemia major, with roughly 150 000 cases documented internationally. At between 10 000 and 15 000 new cases reported each year, India accounts for 25% of the worldwide burden of β-thalassemia. Based on data from the 2011 Census of India, the mean prevalence of the β-thalassemia trait among the 1.21 billion inhabitants of India is estimated to be around 42 million carriers (3–4%) [25]. Population screening has revealed a higher frequency of carrier status in several communities, including Lohanas, Sindhis, Gujaratis, Punjabis, Kolis, Mahars, Bengalis, and tribal groups of Gujarat and Odisha. Even so, the exact number of β-thalassemia traits remains inconclusive. The most affected regions are the western and eastern parts of India. Tribal populations account for higher prevalence of β-thalassemia major, β-thalassemia trait, and other hemoglobinopathies than non-tribal populations. Furthermore, low socioeconomic position is associated with a greater carrier rate of β-thalassemia [26]. Colah et al. report that managing β-thalassemia patients in India poses a significant national health burden [25]. The annual treatment cost for a β-thalassemia major patient is estimated to be between US$629.00 and US$2300.00, with only a few cases fully handled [27]. For example, the cost of transfusing and chelating a child weighing 30 kg was projected to be USD 2985 a year [28]. As the child grows older, a comprehensive approach to therapy is required, which immediately increases the management cost [29]. Several risk factors contributing to the increase of β-thalassemia patients in India have been identified, such as consanguineous marriage (which accounts for 14% of cousin marriages) [30], insufficient public awareness [31], inadequate motivation, high cost of the test, and a dearth of screening infrastructure in the vicinity [32].
India not only faces a large economic burden on the management of thalassemia, but it also deals with SCD [33]. Approximately 120 000 individuals from India were afflicted with SCD in the year 2016 [34]. As per the National Guidelines for Hemoglobinopathies (NHM 2016), the annual expenses for treating SCD, which includes penicillin prophylaxis, are around USD 45 per child. However, this cost does not include the expenditures associated with hospitalization for transfusions and the overall management of SCD [29]. India can reduce the national health burden through the implementation of a number of preventive strategies, such as the development of low-cost molecular diagnostic tests, massive public awareness and education campaigns at all stages of life, emphasizing the significance of prenatal screening and hemoglobinopathies, and the provision of low-cost molecular diagnosis, particularly in rural areas [31], and implementing limitations on the delivery of affected infants at the state and national levels [26].
Progress in molecular techniques in thalassemia diagnosis
Molecular analysis is needed to predict disease severity, from severe, transfusion-dependent thalassemia cases to mild cases that have never been transfused [35] but may have worse survival rates [36], and useful to make accurate diagnoses in cases with complex genetic interactions. There is a plethora of existing molecular techniques that have been used to detect thalassemia, such as reverse dot blot analysis [37, 38], gap-polymerase chain reaction (Gap-PCR) [37, 39], single-tube multiplex amplification refractory mutation system PCR (ARMS-PCR) [40, 41], multiplex ligation-dependent probe amplification (MLPA) [35, 42–44], loop-mediated isothermal amplification (LAMP) [45, 46], Sanger sequencing [47, 48], denaturing gradient gel electrophoresis (DGGE) [49–51], single-strand conformation polymorphism (SSCP) [52, 53], allele-specific PCR (AS-PCR) [54], SNaPshot minisequencing [55], and restriction fragment length polymorphism (RFLP) [49, 56]. As shown in Fig. 1, the legacy techniques based on conventional PCR are laborious, involve multistep analyses and diagnosis, and the decision to choose the panel is based on presumptive diagnosis from complete blood count (CBC) and Hb typing, and thus may lead to misdiagnosis.
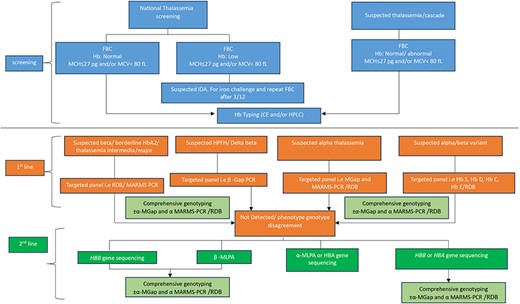
Current workflow for thalassemia screening and diagnosis using conventional PCR (multicenter approach).
Each of these methods has drawbacks, such as long run or hands-on times [57], low resolution, the need for design optimization and/or expensive mutation-specific probes, or the inability to detect point mutations, small mutations, and deletions [58]. Massively parallel sequencing (MPS, aka next-generation sequencing, NGS) has become widely available and integrated by many laboratories in routine genetic disease diagnostics, including thalassemia, to overcome the limitations of existing techniques. Through whole-genome sequencing, exome sequencing, or targeted gene-panel analysis, NGS has helped healthcare workers diagnose and understand complex diseases [59, 60] at a much higher throughput than traditional methods and for many previously undiagnosed disease cases. NGS is now used in many thalassemia diagnostics [61–63]. In 2017, a targeted NGS technique was used to screen 951 minority Chinese Dai people in Yunnan for globin gene cluster carriers [64]. This method detected 49.5% of thalassemia carriers, compared to 22% using conventional methods. NGS is more accurate than conventional methods [65], especially for cases with normal red blood cell indices and hemoglobin fractions that conventional methods may miss [66]. NGS can simultaneously characterize known and unknown deletional and non-deletional mutations in all globin genes [58], reducing the likelihood of false-negative results, misdiagnoses, and the need for additional blood sampling and referral tests for thalassemia screening [67]. Furthermore, the current trend of combining NGS and PCR-based methods is gaining traction among researchers worldwide [63, 65]. Zhang and colleagues screened Chenzhou’s citizens for thalassemia variants using NGS and Gap-PCR in 2019. This combination found rare and novel thalassemia variants [63]. The combined method detects more variants, including rare, common, and novel ones. Thus, it improves at-risk couple detection at a low cost [68]. NGS has many benefits, but it requires expensive equipment and reagents, high sample throughput for pooled sequence analyses, and skilled bioinformaticians [62], whom LMICs lack. The proposed workflow for thalassemia genotyping is presented in Fig. 2.
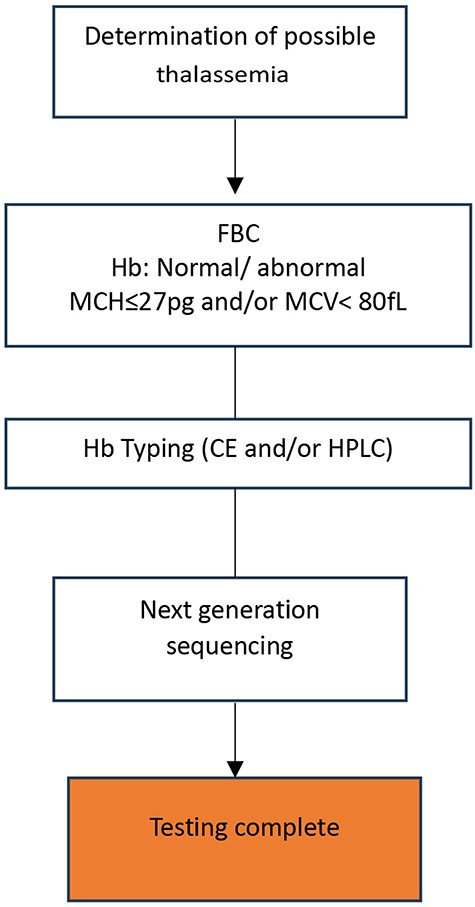
Proposed workflow for thalassemia screening and diagnosis using next-generation sequencing (NGS).
Global globin network
Under the umbrella of the international Human Variome Project [HVP; a UNESCO-partner organization, currently known as Global Variome and now a part of the Human Genome Organization (HUGO)], the Global Globin Network (GGN) is one of the priority initiatives, tackling hemoglobinopathies. The aim of GGN is to improve the quality and quantity of curated genomic data contributed by LMICs to internationally recognized genetic databases, in line with international best practices and in adherence to applicable ethical and regulatory frameworks and policies that assist and protect patients. Additionally, it aims to address the current “digital divide” that exists in the growing field of genomic medicine between HICs and LMICs. Hemoglobin disorders present a significant health problem in parts of the world where genomic technologies are particularly lacking. Thus, they represent an ideal entry point for these countries to develop the genetics and genomic medicine infrastructures and expertise necessary for the detection and reporting of globin variants as a basis for expansion into other areas of health-service delivery.
At the time of this writing, the GGN comprises 32 member countries, made up of 80 individual members including geneticists, hematologists, clinicians, bioinformaticians, and researchers working on hemoglobinopathies. The GGN has been expanded in order to include as many LMIC participants as possible. Of the 32 member countries, 21 are classified as LMICs and 11 as HICs. This composition supports the aim to bridge the marked gap between HICs and LMICs of genomic techniques employed for the diagnosis and management of hemoglobinopathies. The process envisaged to establish optimal disease management across the GGN is to pool knowledge globally, utilize resources synergistically, and integrate advanced diagnostic techniques in health systems to allow evidence-guided implementation of disease management and prevention programs. This is to ensure the achievement of a more uniform and effective approach and outcome, as these will be critically important for those GGN partners projected to experience a growing number of cases over the coming years, which might surpass the capacity of their health system. As a basis for all these efforts, from initial knowledge sharing to long-term training and collaboration, strengthening networking among the GGN partners is imperative. Importantly, since cutting-edge medical genetics and genomics methodologies already exist in most HICs, this can readily be employed to support development and growth in LMICs within the GGN and beyond.
To enlarge the network globally, GGN partnered with several international bodies, such as the Thalassaemia International Federation (TIF), the World Health Organization (WHO), the United Nations Educational, Scientific and Cultural Organization (UNESCO), the ClinGen Hemoglobinopathy Variant Curation Expert Panel [69], and the International Hemoglobinopathy Research Network (INHERENT). INHERENT is the most recent of GGN’s partnerships and sees the GGN participate as a large community within a network of overall over 200 experts from over 110 organizations, spanning 44 countries worldwide, to study genetic modifiers in hemoglobinopathies [70].
Data collection and data sharing
To address the current digital divide amongst LMICs, and between LMICs and HICs, GGN recommended the Leiden Open Variation Database (LOVD) and the ITHANET Portal as platforms for data collection and data sharing. LOVD is designed to support the storage and sharing of genetic and phenotypic information for any kind of disease, so that based on the list of public LOVD installations (https://www.lovd.nl/3.0/public_list; accessed on 17 April 2024), there are currently 4 655 132 variants in 1 803 095 individuals held in 19 LOVD installations worldwide. By contrast, ITHANET (https://www.ithanet.eu/; accessed on 17 April 2024) is a web portal initially conceived specifically for hemoglobinopathies, which, as of this writing, holds in its databases sequence and functional annotation for 3462 variants in 538 genes. Details of 216 experts and 174 organizations working on hemoglobinopathies, epidemiological and healthcare information for 215 countries and regions, and HPLC information for 607 globin variants are also included. More recently, ITHANET launched new databases, namely IthaPhen [71] for genotype-phenotype correlation and IthaCNVs [72] for improved diagnosis of copy number variants in hemoglobinopathies.
Besides geographical epidemiological information, the establishment of databases holding ethnicity information is also important. Knowledge about prevalent variants in individual ethnic groups or communities can be used to streamline diagnostic procedures toward disease prevention and cost-effective health planning. In the area of genotype-phenotype correlation, interpretation of the clinical significance of variants novel to a clinical testing laboratory may be challenging; thus, sharing data among laboratories and standardizing representation are critically important [73]. Therefore, comprehensive coverage of all regions and ethnicities by genomic databases is instrumental for physicians and researchers around the world (i) to obtain information more speedily, (ii) to achieve cost-effective diagnosis and management of genetic disorders, and (iii) to provide improved insight into the causes, severity, and effect of common diseases. In addition, the establishment of, and conclusions derived from, genomic databases increase public awareness of the importance of genetics and genomics as key components of modern health care. This in turn will contribute to public health education as a critical pillar of any successful disease control program.
The importance of establishing a genomic database for thalassemia
The distribution of thalassemia cases among ethnic groups varies according to the variant spectra and their frequencies across different geographical locations, ethnic groups, and population migrations. Thalassemia is an example of a typically recessive Mendelian disorder that is characterized by a distinct spectrum of globin mutations in different ethnic groups. Many of these may also carry characteristic polymorphisms in modifier genes, which are themselves not causative of disease but may at times have a profound impact on disease severity. The enormous diversity of causative β-globin mutations and modifier polymorphisms may be illustrated by using quick filters on ITHANET (www.ithanet.eu, accessed 17 April 2024). Accordingly, for the quick filter “Haemoglobinopathy/β-thalassaemia” on ITHANET, 491 variants were found that affect the HBB gene and cause β-thalassemia (https://www.ithanet.eu/db/ithagenes?action=list&hem=2), whereas a quick filter for “Functionality/Disease modifying mutations” revealed 886 entries, many of which are associated with changes in β-thalassemia disease severity (https://www.ithanet.eu/db/ithagenes?action=list&func=2).
The spectrum of causative and modifier variants may thus differ widely among ethnic groups due to different ancestral variants. Hence, it is necessary to have a database documenting at least all causative variants identified in a specific population, as a vital guide for designing assays and diagnostic tests for targeted detection of variants, and as a resource for the confirmation of diagnoses across diseases with similar symptoms [74].
There is a need for clinicians and scientists to explore variant information using multiple resources, such as publications and databases, to determine whether a suspected “newly discovered variant” has been previously characterized [75]. Ideally, instant access to any variant in a particular gene or locus of interest is required to conduct genomic research efficiently and to deliver “genetic healthcare” to the highest standards [76]. Obtaining up-to-date and accurate information on disease-causing variants is important in the diagnosis of conditions affecting human health. Therefore, a systematic collection of human variants, including genotype–phenotype correlation, is required to assist clinicians and genetic researchers in managing thalassemia globally.
The variants found during clinical testing could form an important regional resource for patient care, especially in LMICs, where information is sometimes not submitted in established databases. This may be due to some clinicians in LMICs being unaware of the existence of corresponding databases. Other reasons are lack of facilities, where laboratories may not be fully equipped to detect variants or may not have the required skills or IT facilities for submission of variants. Beyond technical insufficiency, further impediments to the submission of new variants are a lack of international communications, the absence of DNA bio-banking, restrictions by national authorities, and an inability to translate findings from original languages to English [77].
Existing International Locus-specific Databases on thalassemia affiliated to Global Globin Network
International locus-specific database
Based on various online resources and direct submissions of locus-specific databases (LSDBs), the Locus Specific Database list (https://grenada.lumc.nl/LSDB_list/lsdbs; accessed on 17 April 2024) reports 152 458 public LSDBs. Searching the genes that harbor the clinically most relevant thalassemia mutations, which are HBB, HBA1, and HBA2, several international LSDBs are included in the list. These databases are ClinVar, Global Variome shared LOVD, the Globin Gene Server/HbVar, the Malaysian Node of the Human Variome Project Database (MyHVPDb), and ITHANET. The establishment of international LSDBs is a mark of the success of LSDBs in harmonizing data and training database curators as one of the efforts to increase the quality and usefulness of LSDBs.
ITHANET
The IthaGenes database (https://www.ithanet.eu/db/ithagenes) [78] provides a graphical user interface with live and advanced search functionality and integrates and links gene and variant entries with other data on the ITHANET Portal and elsewhere. IthaGenes holds the most comprehensive dataset of mutations, deletions and copy number variations affecting the globin loci, with 2490 globin mutations (17 April 2024). However, the database also stores 886 disease-modifying mutations in 477 non-globin loci and 90 diagnostically relevant neutral polymorphisms, in acknowledgment of their diagnostic, prognostic, and therapeutic importance for hemoglobinopathies. Where appropriate, such as for epidemiological or chromatography data, data are also rendered graphically to aid comprehension and interpretation [78].
HbVar: a database of human hemoglobin variants and thalassemias
The HbVar database (https://globin.bx.psu.edu/hbvar/) was developed in 2001 by a multi-center academic effort and provides information on sequence variants for the globin genes and related thalassemias and hemoglobinopathies [79–81]. HbVar is a text-based locus-specific database with a total of 1873 database entries for globin mutations (17 April 2024), and with reference to an additional 37 modifier loci through a companion LOVD installation.
ClinGen & ClinVar
In 2013, a Clinical Genomic Resource (ClinGen) (www.clinicalgenome.org), a program under the auspices of the National Institutes of Health (NIH, USA), was launched. ClinGen was established as an authoritative central resource that defines the clinical relevance of genomic variants for use in precision medicine and research. The ClinVar database (www.ncbi.nlm.nih.gov/clinvar) is one of the first resources that was developed for ClinGen and as the primary site for deposition and retrieval of variant data and annotations provides freely accessible reports and supporting evidence linking somatic variants with clinical phenotypes [73, 82, 83]. It is maintained by the National Centre for Biotechnology Information (NCBI) and is supported by intramural National Institutes of Health (NIH) funding [73]. ClinVar is the present gold standard for variant databases, with a steadily growing coverage of variants and with emphasis on the reliability of reported disease association. The proposed thalassemia database will link to corresponding ClinVar entries, where these are available. Variants that are reported in ClinVar are determined by clinical testing laboratories, in contrast to the Human Gene Mutation Database (HGMD®) [84] and OMIM (www.omics.org/), which focus on variants documented in the literature [85]. Also, the variants reported in ClinVar include both germline and somatic variants, while COSMIC includes only somatic variants [86].
The Malaysian node of the human variome project database (MyHVPDb)
The Malaysian Node of the Human Variome Project Database (MyHVPDb, https://myhvpdb.kk.usm.my/genes; accessed on 17 April 2024) is an LSDB that stores human variants and phenotypes specific to Malaysia [87]. MyHVPDb was established in 2011 and collects, displays, and curates DNA variants in specific genes and phenotypes found in the Malaysian population, with emphasis on the genetic heterogeneity of the Malaysian ethnic groups. Based on LOVD 3.0 [88], MyHVPDb now integrates 3734 variants from 311 genes and 165 diseases, as collected from various databases, including PubMed, Scopus, and Google Scholar. MyHVPDb stores information on summary variant data, as well as full case-level information on individuals, phenotypes, screenings, and variants, with current limitation of clinical parameters to thalassemia disease. MyHVPDb has been configured to appear in the global listing of the LOVD central server in order to allow global data sharing and as a critical tool for data-guided disease management in Malaysia and in Malaysian or ethnically similar communities elsewhere.
The case for multiple hemoglobinopathy data resources
ClinGen/ClinVar are the most reliable, genome-wide annotated resource for knowledge about disease causation by genetic variants. For a quickly expanding diagnostic landscape based on NGS, reliable annotation for the evaluation of variants detected across the genome is essential. However, this resource does not consider differences in disease severity or different phenotypic features for syndromic disorders, which is essential for specialists working on specific diseases, and it does not (currently) consider the influence of modifiers.
ITHANET with IthaGenes is the presently most comprehensive resource for hemoglobinopathies, which, beyond variant information for causative genes, also includes modifiers, detailed information on phenotypic features, detailed epidemiological information, national health policy and management information, and advanced tools for genotype-phenotype prediction for genetic counseling. For specialists working on hemoglobinopathies, this is an unrivalled and openly accessible resource. On the downside, it is not suitable for uncurated submissions and local installation for offline access.
LOVD is open-source, can be freely and easily installed locally in any environment (including environments off the grid or behind stringent firewalls), is widely used for many individual, LSDBs, is supported by frequent workshops, e.g. at the European Society of Human Genetics (ESHG) and HUGO meetings, and even allows installation of individual databases on a server in Leiden, the Netherlands, for those who cannot operate their own database server. It allows the storage of information for individual patients as well as automatic import of NGS data and synchronization with other LOVD databases. While the platform has limited support for same-disease information across loci, epidemiological information or integration of graphical information, such as HPLC chromatograms or maps, which are of great utility for hemoglobinopathies, it is an excellent choice for local database installations in low-resource environments, which nevertheless allows harmonization and synchronization with centralized databases. For this reason, it has been the choice for the Malaysian Node for the Human Variome Project Database and is the suggested platform for a central thalassemia database.
Curation of the database using the LOVD platform
We recommend using LOVD 3 as it has offered many advantages for curators to manage LSDBs. This includes its flexibility for customization and extension, thus enabling the identification and addition of data items required for the extraction of a wide range of information at the user end.
LOVD 3 also offers five different access levels, i.e. administrator, manager, curator, submitter, and general user. Based on multiple access levels, it is easier to draw on a team of curators or expert advisors, which in turn facilitates data collection, verification, and submission from various centers and for dissemination of information to the general public. Furthermore, if necessary, the LOVD 3 system allows retrieval and transfer of the entire dataset to another suitable web-based application [89].
LOVD 3 was developed to connect with various resources, such as HGNC, NCBI, EBI, and Mutalyzer, as a basis for ongoing high-quality data provision. For instance, LOVD may draw on the NCBI sequence viewer to provide a visual representation of each database entry at its corresponding location and its interconnection with other NCBI resources [90]. The NCBI sequence viewer was selected over other genome browsers because of its simplicity and the ability to embed it in any page without the need for local installation and administration [91]. Moreover, LOVD allows links to original publications using the PubMed ID and/or DOI.
LOVD 3 provides different tables and linked tables, including individual, phenotype, screening, and variant(s). The “individual” table contains details on the family/patient studied, including gender, geographic origin, and patient identification as listed in the original publication. For thalassemia as a disease, the “phenotype” table includes the disease phenotypes as originally reported, which, in the case of the MyHVPDb, consists of clinical phenotype, clinical severity, age at thalassemia presentation, age at receiving the first blood transfusion, growth and development, hemoglobin level (g/l), the requirement for blood transfusion and spleen size (cm), all of which may be added to the database according to the clinical parameters of β-thalassemia patients. The “screening” table contains the detection template (DNA/RNA/Protein), techniques used, and genes screened. Meanwhile, the “variants” table includes information on allele, chromosome, genomic DNA change, published as, reference, dbSNP ID, frequency, and affects function.
It is essential to evaluate data quality during data collection, because entries may affect health decision-making, research, and clinical practice [92], and because the quality of content may differ widely depending on the responsible curator or contributor. Therefore, LOVD 3 was designed to fulfill this requirement, which may be one of the reasons why the customizable platform is a frequent point of reference for the Working Group for Variant Database Quality Assessment Criteria.
Guidelines for genomic databases for thalassemia
This paper aims to provide the minimum requirements to enhance the contents of existing genomic databases in thalassemia that are based on the LOVD 3 platform (Supplementary Table S2). Therefore, it is well recognized that genomic databases for the annotation of genetic variants and phenotypes for thalassemia have great utilitarian value [92]; however, their inconsistencies across institutions (and more so countries) are of concern. Although not all the necessary information may be available for variants previously published in the literature, databases should at least be designed to include minimally required content. Figure 3 depicts an example of a genomic database with suggested minimal reporting of information for each case and variant observed, placing focus on material that can be accessed by the general public, while hiding more specific information that is restricted from access [93]. Around minimal content and structures for genomic databases, additional points need to be laid out in order to harmonize and facilitate ongoing contribution by multiple users, including terms and definitions, nomenclature and standards system, optional content and structures, and guidance on implementation with the database management software in question (Fig. 3). Adherence to these guidelines allows for usability but also great flexibility in the implementation of different databases of relevance for hemoglobinopathies, as listed in Table 1.
![An overview of five key elements as the main core needed for the establishment of the thalassemia database. Adapted from [93].](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/database/2024/10.1093_database_baae080/1/m_baae080f3.jpeg?Expires=1788392049&Signature=EfYXiu3R7eVwMLJIqNWl8NkouuYh5h6hoIIP38~~3CLDaH-Q-hZgOIi6WyZil1uliy8oUHBdhmsvJ2LsdSxoDfDtA7-Kqo0uUQ3wj2z0M4xzJXAZnJltzbu4cicqizSvYO-b9E2V-RZb94e-2LytD7BuWttiKY7BZ-rY4BUN6RqHJ5n5Fo-jBEexU5Mdx78tt-1vPbljY6tKwiOlsJKSYlmg1E9W8NQECoZ5abjR8YRGWl9NzK9HnqdWRQ6D6HiC2zthP11FSZP2hZsidIx8j90Xsdes9s0TbVuicHR811baFq6XbbyZsd7-XmZ37630egRPDOBBNJJ8PvUhps0CFw__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
An overview of five key elements as the main core needed for the establishment of the thalassemia database. Adapted from [93].
Databases of thalassemia around the world
| Databases | Diseases/Description | Link | References |
|---|---|---|---|
| ClinVar | Genetic diseases including thalassemia | https://www.ncbi.nlm.nih.gov/clinvar/ | [73] |
| HbVar | Information about hemoglobin variants and mutations that cause thalassemia | https://globin.bx.psu.edu/hbvar/ | [81] |
| Malaysian Node of the Human Variome Project Database (MyHVPDb) | Genetic diseases including thalassemia | https://myhvpdb.kk.usm.my | [87] |
| ITHANET | Thalassemia Sickle cell disease | https://www.ithanet.eu/ | [106] |
Terms and definitions
The description of genomic data (variant- and case-level data), variant classification, assertion (of pathogenicity or otherwise), clinical significance, functional significance, attributes, and acceptance values are listed in the section of terms and definitions in the Supplementary Materials.
Nomenclature and standards systems classification
As the required nomenclature system, gene names and variant descriptions ought to follow guidelines of the HUGO Gene Nomenclature Committee (HGNC) (https://www.genenames.org/) and HGVS Sequence Variant Nomenclature [94], respectively. Likewise, phenotype descriptions should consistently follow terms specified by Human Phenotype Ontology (HPO) (http://human-phenotype-ontology.github.io/), International Classification of Diseases (ICD, http://www.who.int/classifications/icd/en/), or International Classification of Diseases for Oncology (ICD-O, http://codes.iarc.fr/), depending on the focus of the database. However, the Online Mendelian Inheritance in Man (OMIM) (https://www.omim.org/) is a database used to define diseases. OMIM should be included with HPO in a genomic database to complement the information about the disease and phenotype [95]. For disease ontologies, guidelines from Orphanet Rare Disease Ontology (ORDO) [96], Mondo Disease Ontology (MONDO) [97], and Dictionary of Disease Ontologies (DODO) [98] are recommended. Besides, for recommended standards, a number of variant classification standards have been created, as detailed under Terms and Definitions.
Guidelines for minimal contents
Depending on the deployed portal or database system, the minimally required contents can be separated into two distinct categories: database-level information and record-level information (Fig. 3).
Mandatory database-level information
The following details must be presented on the website for the database, as detailed elsewhere [99, 100]. For thalassemia genomic variant databases per gene, there are a few vital elements that should be focused on, including the use of HGNC Gene symbols, HGNC IDs, OMIM, and HGNC-approved names. Reference sequences of the genome must be based on a build of the genome that is up-to-date and well-defined (please refer to http://varnomen.hgvs.org for details). In other circumstances, suitable RefSeq, chromosomal/genomic, transcript, and protein sequences should be referenced [101]. Any deviation from reference should be described in detail, and the reference sequence used for variant descriptions in the database should be specified. This information is generally available in LOVD via the RefSeq URL link on the gene homepage. Lastly, a specification for multivariant allele and/or aberrant sequence variant nomenclature may be added in addition to the obligatory HGVS sequence variant nomenclature. Any relevant information on imprinting, which influences gene expression and thus illness or trait inheritance, should be presented. Such information helps prevent problems in the interpretation of RNA-based studies, such as mistaken assumptions of homozygosity or difficulties explaining the absence of DNA-level variations at the RNA level. As additional details, information and/or stable connections to other online resources (such as GeneReviews; https://www.ncbi.nlm.nih.gov/books/NBK1116/ and Genetics Home Reference; https://ghr.nlm.nih.gov/) provide access to further biological and medical information that may be valuable to the user community.
Mandatory record-level information
Mandatory variant information (variant-level data) and mandatory individual information (case-level data) are the minimally required data for genomic variant databases and both are vital to reporting variations and phenotypes.
Mandatory variant information (variant-level data)
For the mandatory variant information (variant-level data), there are a few elements of key concern. First, high-throughput sequencing (next-generation sequencing, NGS) is becoming increasingly important in the field of DNA diagnostics and extends beyond individual loci, so that variants need to be described at the chromosomal level, for example, NC_000023.11:g.115641445_115653674del. Researchers from LMICs can benefit from NGS and gain experience with the platform through collaboration with their counterparts in HICs, which might take the form of academic collaboration or, more controversially, of business investment in future markets for diagnostic and medical products. The increased disease burden in HICs by migration from LMICs may also create incentives long-term for HICs to invest in training, funding, and logistics support for NGS in LMICs, similar to what is being proposed for the control of other global health challenges, such as antibiotic resistance [102]. Because of this, bioinformatics pipelines can leverage HGVS genomic DNA variant descriptions, which are very straightforward, in database searches and (automated) submissions. Using chromosomal-level descriptions eliminates errors caused by improper HGVS coding DNA sequence variant nomenclature in NGS pipeline variant callers. The description of every coding DNA sequence on a chromosome is redundant from the genomics perspective, which concentrates on chromosomal-level descriptions. In addition, at the level of the RNA transcript, a variant is described as an example, e.g. NM_003001.3:r. = (no effect), NM_003001.3:r.(23dup)(predicted), NM_003001.3:r.23dup (RNA analyzed). When describing a variant at the RNA and coding DNA levels, the same transcript reference sequence is used, but with a different prefix. The HGVS nomenclature demands predicted consequences have to be reported in parentheses “()” as an example; NM_003001.3:r.(23dup). The goal of describing a variant at the RNA level is to show how the variant located at the coding DNA level is projected or spotted to modify the resulting transcript. It is important to report all possible products resulting from the partial effects on splicing, whether they have been examined or prognosticated. Furthermore, the variant at the coding DNA level [e.g. NM_003001.3:c.(23dup)] is described using the transcript reference sequence in compliance with HGVS nomenclature criteria (see http://varnomen.hgvs.org/bg-material/refseq/). The MANE transcript for a gene should always be used for the reporting of variants on the transcript level. When variants are reported in relation to a transcript, the preferred reference sequence is the reference suggested by the Matched Annotation from the NCBI and EMBL-EBI (MANE) project. The main goal of this explanation is to raise awareness regarding the possibility of incorrectly interpreting variant effects at the transcript and protein levels due to aberrant splicing caused by the observed variant. At the level of the protein, a variant is described as follows: NP_002992.1, e.g. NP_002992.1:p.(His8Glnfs*12). The parental origin of the variant also needs to be described. This information is critical for both recessive and imprinted disorders, as well as for the evaluation of de novo variants in autosomal dominant disorders. Techniques such as SEQ (sequencing), MLPA (multiplex ligation-dependent probe amplification), SEQ-NG (NGS), PTT (protein truncation test), and array SNP are used to detect and validate the sequence variant. In addition, all molecule types (unknown, DNA, RNA, or protein) used to detect or confirm the reported variation instance should also be specified. This information and the methodologies employed are intended to facilitate data quality assessment, while also providing other institutions with guidance when implementing similar testing/investigations of their patients. Finally, each variant should be assigned a unique and permanent identifier, to allow unambiguous reference, and in turn a link to the original information source should be provided, such as a publication, submitter, or central database.
Mandatory individual information (case-level data)
Mandatory individual information (case-level data) includes a unique submitter ID (preferably ORCID) and a pseudonymized, potentially non-public lab-ID code for each analyzed individual, so that the individual cannot readily be identified from the data. Each case-level record in the database also needs to have a consistent, unique, and permanent identifier. Besides, for individuals with a disease phenotype, the phenotype is reported, whereas for pre-symptomatic or carrier individuals the phenotype of an affected family member is reported; additionally, the genotype of unaffected family members is given. In addition, the list of potential genes (for a certain disease or trait) that have been evaluated in a person, without detecting variations, should also be reported. Importantly, case-level data will be essential for the recording of phenotypic variability typical of hemoglobinopathies. It will also record any variants detected in known disease modifiers to allow an analysis of their influence on the phenotype across databases/across all case-level data in the centralized database.
Recommended record-level information
It is recommended that the contents of genetic variant and disease-specific databases be expanded as described below for a more comprehensive evaluation of the clinical effects of variants. Additionally, the specified fields and their contents for genetic variation databases can be classified into variant-level and case-level data.
Recommended variant information (variant-level data)
Several details should be highlighted for the recommended variant information (variant-level data). First, the variant at the level of gene- or region-specific DNA needs to be described, regarding which one can refer to the HGVS sequence variant nomenclature (http://varnomen.hgvs.org/) for more information, permitted values, and further examples. The functional consequence (functional significance) such as descriptions: not known to affect function (-), probably not affecting function (-?), unknown (?), probably affects function (+?), affects function (+), needs to be included as well. Ideally, these descriptions are accompanied by a reference to the source of the claim (submitter, curator, expert panel, functional database, PubMed ID). Besides, the number of times the variant allele was reported is also important to include, especially for variants that have functional effects [such as 1 = found in one patient only, 12 = found in 12 individuals (patient and other family members)]. It is recommended to include the number of probands separately from family members. It is also necessary to provide information about the genetic origin of the variant, such as germline, somatic, de novo, (unspecified, maternal, or paternal) uniparental disomy, and in vitro (cloned for functional testing). Additionally, it is critical to include information about the tissue sample(s) used to find the variant, comments on the described variant, links to population frequency databases, the number of exons/introns affected by the variation, and the frequency with which the variant allele is reported. Furthermore, the description of the variant as it might have appeared in the original publication, its common name/alias, and the phenotypic segregation of the variation are also important. Variant-phenotype segregations, such as descriptions: ? = unknown, yes = segregates with the disease, no = does not segregate with the disease, even more significant if providing the number of segregations or non-segregations, exon and intron numbers affected by the variation (as an example, 01 or 01e = exon 1, 02i = intron 2, 03_07 = exons 3 to 7) and links to databases of population frequency, are also crucial to introduce in the variant-level data section. Lastly, the assertion needs to be provided. For further information, please refer to the section on terms and definitions in the supplementary materials. If different variants cause different diseases, the clinical assertion should also include the disease against which the assertion is made. It would be preferable to include a description of the evidence used in the clinical assertion of the variant, as well as references.
Recommended individual information (case-level data)
There are a few important components that should be considered for the recommended individual information (case-level data). First, the disease’s name in other databases or elsewhere needs to be mentioned. A list of the genes tested in the individual also needs to be included. In the case of exome sequencing, for example, a link or reference to the list of genes included in a capture kit is sufficient. It is also worth mentioning whole-genome sequencing, if done. The details such as age at diagnosis, gender of the person, consanguinity of the parents’ marriages, the number of patients reported in a family or population, the individual’s geographical/ancestral origin, remarks about the person, extended phenotypic information about the person tested, and the inheritance pattern of the disease or trait in the individual’s family should be included. Finally, the name/alias of the disease in other databases should be included, and the individual population should preferably be based on self-assessed indigenous groups as well.
Optional database contents and structure
Several recommendations could be explored to enhance the utility of databases:
Storing variant combinations (alleles and genotypes) observed in each gene (investigated) per individual. In theory, databases can employ the HGVS allele or genotype description format; however, this is rarely done to avoid variant search and sort issues. This includes linking all cis or trans variants found in the individual’s record—combining this information with each individual’s health or disease state will support key conclusions about the impact of variations, especially those responsible for recessive disorders or detected in critical genes or those that may modify the phenotype.
Storing all gene variant data from an individual, especially variants of unclear significance, allows for frequency data collection, which aids in classification judgments (or even future analysis pertinent to genetic modifiers of the disease phenotype).
Comments should be enabled, with ID tracking for submissions and edits by registered users, who after all are the experts in the topic and are frequently the database curators.
The addition of functional assays and detailed phenotypes to genetic variation databases may transform them into knowledge bases, given the availability of appropriate search and interpretation tools.
It is anticipated that links to precise phenotypes, pedigree information, time course changes, and other identifiable information in specialized patient and disease registries with minimally restricted access regulations will boost the clinical usefulness of these databases.
Implementation of minimal, recommended, and optional content
In genomic variant databases that use the LOVD platform (https://www.lovd.nl/), additional information may be found about the contents of the various database fields and examples of how they may be implemented or expanded (Fig. 3). We also propose the list of informative phenotypic data for thalassemia patients, which can be managed in LOVD, by referring to Supplementary Table S1.
Toward an instance of the thalassemia database
From concept to concrete database, the GGN and the Malaysian HVP node have been co-organizing workshops for the implementation of LOVD-based local databases for hemoglobinopathies and their connection to central database installations in Leiden. As GGN member countries are making efforts to achieve funding for such implementations, education continues to cover essential practical, safety, ethical, and quality aspects for the envisaged database. This includes considerations for the establishment of (local) database instances, such as that it be non-redundant with existing resources, that long-term funding is available for maintenance, that a common data model is applied, allowing exchange of data, and that its data can readily contribute by synchronization or other means to the centralized Thalassemia database [103]. Tied in with this are aspects of quality for each database, which goes from (i) data quality, e.g. by measures to reduce clerical errors and ensure accountability, over (ii) technical quality relating to the quality of the platform, control measures, and speed, over (iii) timeliness concerning frequent data updates, to (iv) accessibility, which includes adequate documentation, searchability, and usability of the platform interface [80]. Such training further covers the awareness and adherence to high curation standards to turn each local database and thus the central resource into a meaningful tool for the community [104], and likewise awareness and means of maintaining confidentiality and privacy of data through suitable security and pseudonymization procedures in local storage and in the national and international sharing of data [105].
Conclusion and recommendations
In 2015, the Human Variome Project, a worldwide non-governmental organization, launched GGN with a focus on thalassemias and hemoglobinopathies. Since then, the GGN has grown rapidly and attracted many participants from LMICs endemic for these disorders. Its focus is on hemoglobinopathies as a gateway to genomic medicine in low-resource environments and as a proof of concept for the case of international sharing of hemoglobin variant information, enabling expertise and knowledge sharing among the involved countries and spurring a variety of research projects in the field. One of its major initiatives is spearheading the creation of global thalassemia and hemoglobinopathy databases and resolving the challenging technical aspects of its development, while maintaining close collaboration with existing regional or country-based databases. This database has an additional benefit over heterogeneous patient-centric registries, in that the data are anonymous, allowing for more extensive worldwide data exchange with fewer restrictions for various clinical and academic applications. The database is also aligned with the variant classification using the ACMG/AMP guidelines. Additionally, it has identified a collaborative network of experts who are eager to participate in the different facets of variant sharing between nations and who are sensitive to meeting the needs of diverse populations. The establishment of individual LOVD-based variant databases in GGN member countries and their integration with the ITHANET Portal as a central resource for hemoglobinopathies as part of this effort may be instrumental in initiating essential data-guided disease management in LMICs and in capturing the diversity of hemoglobinopathies as an international resource.
Acknowledgements
We would like to acknowledge the contributions of the late Prof Zarina Abdul Latiff and the late Prof Amer Feisal Merican, who passed away while preparing this manuscript. Please see the supplementary materials for the list of members of the Global Globin Network (GGN).
Author contributions
CWL, CB, PK, YH, AFM, MHM, NFMY, EE, JE, DC, RZARS, GEK, MKT, NMY, ZAL, RR, and BAZ: conceptualization and writing—review and editing. HHF, NNZ, and HA: writing—original draft preparation. NNZ: visualization. BAZ, NMY, and ZAL: supervision. MK, BAZ, CWL, PK, NMY, ZAL, and JB: funding acquisition. All authors have read and agreed to the published version of the manuscript.
Supplementary data
Supplementary data is available at Database online.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was funded by the Ministry of International Trade and Industry (MITI), Malaysia, 304.PPSP.6150166.K151; the International Collaboration Fund (IFC), Ministry of Science, Technology and Innovation (MOSTI), Malaysia, 305.PPSP.6114202. This work was co-financed by the European Regional Development Fund and the Republic of Cyprus through the Research and Innovation Foundation (Projects: EXCELLENCE/1216/92, EXCELLENCE/1216/256). This work was facilitated by COST Action CA22119 (HELIOS), supported by COST (European Cooperation in Science and Technology).
References
Author notes
contributed equally to this work and share first authorship.
The full membership listing of GGN is given at the supplementary materials of this article.

{kind=link}
{kind=link}
{kind=link}