





Abstract
The identification of chemical–protein interactions described in the literature is an important task with applications in drug design, precision medicine and biotechnology. Manual extraction of such relationships from the biomedical literature is costly and often prohibitively time-consuming. The BioCreative VII DrugProt shared task provides a benchmark for methods for the automated extraction of chemical–protein relations from scientific text. Here we describe our contribution to the shared task and report on the achieved results. We define the task as a relation classification problem, which we approach with pretrained transformer language models. Upon this basic architecture, we experiment with utilizing textual and embedded side information from knowledge bases as well as additional training data to improve extraction performance. We perform a comprehensive evaluation of the proposed model and the individual extensions including an extensive hyperparameter search leading to 2647 different runs. We find that ensembling and choosing the right pretrained language model are crucial for optimal performance, whereas adding additional data and embedded side information did not improve results. Our best model is based on an ensemble of 10 pretrained transformers and additional textual descriptions of chemicals taken from the Comparative Toxicogenomics Database. The model reaches an F1 score of 79.73% on the hidden DrugProt test set and achieves the first rank out of 107 submitted runs in the official evaluation.
Database URL: https://github.com/leonweber/drugprot
Introduction
With the rapid growth of biomedical literature, it is becoming increasingly difficult to obtain comprehensive information on any entity, such as a specific gene or drug, by only reading. Important aspects of biomedical entities are their interactions with other biomedical concepts. This is especially true for the relations between drugs and proteins, which are of high importance in various applications such as drug discovery (1), precision medicine (2) and curation of biomedical databases (3). Manual extraction of such relationships from the biomedical literature is costly and often prohibitively time-consuming. As an alternative, information extraction can help to automatically identify these relationships at a large scale and make them more readily accessible. Accordingly, extracting (biomedical) relationships from text has been investigated intensely over the last two decades (4). These methods generally employed hand-crafted features based on lexical or syntactic information (5), kernel-based learning (6) or various forms of neural networks (7). Moreover, combinations of the approaches have been applied (8).
Most recently, a variety of methods employed pretrained (transformer-based) language models and achieved new state-of-the-art performance across several domains and information extraction tasks (9, 10). The language models are trained without supervision on large unlabeled text corpora (e.g. Wikipedia articles or PubMed abstracts) first and then fine-tuned to one (or more) target tasks, e.g. named entity recognition (11), relation extraction (10) or question answering (12). A body of work has addressed the assessment of such language models on biomedical texts and made their models publicly available for further research (10, 13).
One of the challenges of machine and deep learning based models is that they typically require large amounts of labeled data (14). However, in some specific domains, e.g. in biomedicine or materials science, the amount of labeled data available is low and precisely annotating texts is difficult, since this requires expert knowledge and a lot of time (15). In order to address this problem, a plethora of approaches explored methods of enriching and augmenting the little data available (16–18). Recent studies explore different transformations of existing instances without changing their label, e.g. via synonym replacement (16), switching (uninformative) words (18) or back-translation (17), to build additional training data. For instance, Wang and Henao (17) use pretrained machine translation models for generating paraphrased sentences to improve named entity recognition models in low-resource settings. Wei and Zou (16) replace words with one of their synonyms retrieved from a thesaurus (e.g. WordNet). Kobayashi (19) substitutes words with words predicted by a language model at this position. Moreover, researchers experimented with incorporating additional side information from domain-specific knowledge bases (KBs) to enhance their models (20, 21). For example, Vashishth (20) propose a distantly supervised method, which applies Graph Convolution Networks to encode syntactic information from text and utilizes additional KB data for improved relation extraction. Xu and Barbosa (21) describes a framework for joint training of heterogeneous representations of text and from facts in a database using KB embeddings (KBEs).
A common use case for relation extraction models is KB population (KBP), in which the model is used to extract relations from a large collection of texts (7, 22–25). Then, the resulting relations are added to a KB, possibly after undergoing manual curation or automatic plausibility checking. For instance, Ernst et al. (26) introduce KnowLife, a large KB for health and life sciences, automatically constructed from scientific publications, health portals and online communities using a small number of seed facts in a pattern-based, distantly supervised approach (27). Moreover, they use confidence statistics and logical reasoning for consistency constraint checking to achieve high precision of the identified relations. Singhal et al. (28) propose a machine learning approach to curate a biomedical KB for precision medicine via extracting disease-gene-variant triplets from biomedical literature. In contrast, Weber et al. (7) combine deep language models and distant supervision for identifying functional protein–protein associations as well as text spans stating the associations in the literature. An expert evaluation highlights that the approach is able to extract protein–protein relations that are missing from major pathway databases.
Since 2003, the BioCreative initiative organizes challenges to foster the development and evaluation of text-mining approaches in the biomedical domain. In 2016, they hosted a first shared task on chemical–protein relation extraction (29). Track 1 (DrugProt) of the 2021 BioCreative VII challenge (30) explores the recognition of chemical–protein relations in scientific abstracts. The organizers compiled a manually annotated corpus of abstracts labeled with all chemicals and gene/protein mentions as well as binary relationships between them, categorized into 13 different types of interactions. Participants of the challenge were asked to develop methods which, given the abstract text and annotations of the mentioned chemicals and proteins, detect all binary relations and their type.
In this paper, we describe our contribution to this challenge. We define the task as a sentence-level relation classification problem, i.e. given a sentence and all chemical–protein pairs mentioned in it, for each pair to predict the type of relationship they are in (or ‘no’ as a special type). Our approach is based on pretrained transformer-based language models. We investigate the extension of this relation-classification baseline model by adding textual and embedded side information from biomedical KBs. Moreover, we explore the effect of increasing the size of training data by using an additional gold standard corpus as well as generating paraphrased instances via back-translation. We perform a comprehensive evaluation of the proposed model and the individual extensions including an extensive hyperparameter search leading to 2647 different runs.
Our best model is based on an ensemble of 10 pretrained transformers and additional textual definitions of chemicals taken from the Comparative Toxicogenomics Database (CTD) database. The model achieves an F1 score of 79.73% on the hidden DrugProt test set and achieves the first rank out of 107 submitted runs in the official shared task evaluation. Furthermore, our experimental results highlight the necessity of extensive hyperparameter tuning to reach state-of-the-art extraction performance. Our code and model are publicly available (https://github.com/leonweber/drugprot).
Materials and methods
Task and datasets
For the DrugProt shared task (30), the organizers provided a data set of 4250 PubMed abstracts with gold standard annotations for gene/protein and chemical mentions, as well as for relations between them. The goal of the shared task was to use these abstracts to build a system that can accurately detect and classify chemical–protein relations in biomedical text. The participating systems were evaluated on another set of 750 abstracts for which gene/protein and chemical mentions were provided but the chemical–protein relations were hidden to ensure a fair evaluation. Chemical–protein relations are labeled with one or more of 12 relation classes. Detailed data set statistics can be found in Table 1.
Document, entity and relation statistics of the DrugProt data set.
| Train | Dev | Test | |
|---|---|---|---|
| Abstracts/Passages | 3500 | 750 | 750 |
| Chemicals | 46,274 | 9853 | 9434 |
| Genes/Proteins | 43,255 | 9005 | 9515 |
| Total | 89,529 | 18,858 | 18,949 |
| Activator | 1428 | 246 | 334 |
| Agonist | 658 | 131 | 101 |
| Agonist-Activator | 29 | 10 | 0 |
| Agonist-Inhibitor | 13 | 2 | 3 |
| Antagonist | 972 | 218 | 154 |
| Direct-Regulator | 2247 | 458 | 429 |
| Indirect-Downregulator | 1329 | 332 | 304 |
| Indirect-Upregulator | 1378 | 302 | 277 |
| Inhibitor | 5388 | 1150 | 1051 |
| Part-Of | 885 | 257 | 228 |
| Product-Of | 920 | 158 | 181 |
| Substrate | 2003 | 494 | 419 |
| Substrate_Product-Of | 24 | 3 | 10 |
| Total | 17,274 | 3761 | 3491 |
We experiment with multiple model modifications which require linking the entity mentions to reference ontologies. We link mentions of chemicals to the CTD chemical vocabulary (http://ctdbase.org) (31), which provides Medical Subject Headings (https://www.nlm.nih.gov/mesh/meshhome.html) unique identifiers, while we link mentions tagged as genes/proteins to the National Center for Biotechnology Information Gene database (https://www.ncbi.nlm.nih.gov/gene) (32). To perform the normalization, we employ BioSyn (33): a state-of-the-art dense neural retrieval model using BioBERT (10) as the backbone pretrained language model. We train a normalization model for chemicals on the entire BioCreative V CDR (BC5CDR) dataset (34) (train+dev+test) and on BioCreative II Gene Normalization (BC2GN) (35) (train+test) (as provided by (36)) for proteins. We use the author’s original implementation (https://github.com/dmis-lab/BioSyn) and train the models for 20 epochs with the Adam optimizer (37). For all other hyperparameters we use the values suggested by BioSyn authors. At inference time, the models encode both all names in the given ontology and the mentions to be normalized in an embedding space and select the candidate with the highest inner product score for each mention. An estimate of the accuracy of these models can be found in (36), who find that the model achieves 83.8% accuracy for unseen chemicals and 85.5% for unseen genes when trained on a subset of our training data.
Base model
We now report the evaluated model configurations by first outlining the baseline and then describing all tested modifications. We highlight the hyperparameters and modifications of the best performing model (as measured in F1 on the DrugProt test set) in bold.
We frame chemical–protein relation extraction as a multilabel relation classification problem, which we approach by finetuning pretrained transformers. More specifically, we generate one training/testing example per pair of entities that occur together in the same sentence. We insert the special tokens [CLS], [HEAD-S], [HEAD-E], [TAIL-S] and [TAIL-E] into the sentence, where [CLS] is the classification token prepended to the sentence and the other four mark beginning and end of the chemical (head) and protein (tail) entities, respectively. See Figure 1 for an example. We also experiment with masking the head and tail entities by replacing them with HEAD and TAIL to prevent the model from associating specific pairs with relations without taking the context into account. Then, we use a pretrained transformer to obtain a contextualized embedding hi of every token in the sentence and represent the sentence by using the embedding of the [CLS] token.
![Overview of our base model and all evaluated extensions. Solid lines indicate the components of the base model, whereas dashed lines indicate evaluated extensions. We create one input example per chemical–protein pair in each sentence and mark the pair with special tokens. This sentence is embedded with a pretrained language model. Finally, the embedding of the [CLS] special token is passed through an output layer. As pretrained language model, we use RoBERTa-large-p.m.-M3-Voc in our base model and evaluate replacing it with four other variants. Textual information is appended to the input text, and KBEs are concatenated with the [CLS] embedding.](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/database/2022/10.1093_database_baac098/1/m_baac098f1.jpeg?Expires=1787635559&Signature=LezDuBxmRIIdEfZW0rd95cy-eEx6GK-ZvmOG9v6RDxHpx~tD~26G9uczgPMU-2SSz1sfMhhNPR~CjYfvRhqDumfpG30DJZtuQgs2xVYRC-BmB87bRU9tWRZBPs30CV9HgM-PHv1B5av76LCuRgFoqNjUXVvgbyySiTulMx2E4kbxux2FW9phsoS9EVxq5Pa5esvfU5~3d5ooY5Vn-t2KIhvkU~~scEDb64HByLCK0S1lAYqiYCCOBZcS3Q8brDZLXPuiGE3f6kxevglX0uDWp1jjlSwFY-rtQKrJ5Cu39WdOZ4lu6juaJHRJCz-xJliUxeVngUB6V-JmUtQNBKx9IQ__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Overview of our base model and all evaluated extensions. Solid lines indicate the components of the base model, whereas dashed lines indicate evaluated extensions. We create one input example per chemical–protein pair in each sentence and mark the pair with special tokens. This sentence is embedded with a pretrained language model. Finally, the embedding of the [CLS] special token is passed through an output layer. As pretrained language model, we use RoBERTa-large-p.m.-M3-Voc in our base model and evaluate replacing it with four other variants. Textual information is appended to the input text, and KBEs are concatenated with the [CLS] embedding.
Finally, we apply a linear layer that maps the sentence representation to the logits, which are then normalized with a sigmoid nonlinearity. To compute the loss we use binary cross entropy. We optimize our model using Adam (37) with a learning rate schedule in which the learning rate is linearly increased from zero to the target learning rate during the first 10% of training steps and then linearly decayed to zero for the remaining 90%. We explored the following hyperparameters for the base model:
learning rate: {5e-6, 3e-5, 5e-5}
epochs: {3, 5, 10}
maximum sequence length: 256
batch size: {8, 16, 32}
Language models: PubMed-BERT-abstracts and PubMed-BERT-abstracts-full-text (13), BioBERT-v1.1 (10), BioMED RoBERTa (38) and RoBERTa-large-PM-M3-Voc (39).
Entity masking: {true, false}
Textual side information
We conjectured that enriching the input with information that augments the sentence context might lead to a more accurate model, for instance that a chemical is known to act as an agonist for a certain class of proteins or that a protein belongs to a specific protein family. To this end, we experiment with different additional textual information concerning chemicals and proteins gathered from different KBs. That is, for an example in which the chemical c and the protein p are marked as head and tail, respectively, we queried a database for textual information on c, p and appended this information to the input. See Figure 1 for an example. In cases where this led to a number of tokens exceeding the maximum sequence length, we first truncated the side information before truncating the input sentence. When the query for chemical side information did not yield any results, we instead searched for side information on the chemical’s parent in the hierarchy of the CTD database’s (31) chemicals vocabulary (http://ctdbase.org/reports/CTD_chemicals.csv.gz). Specifically, we explored the following choices for textual side information:
ChemicalDefinition: The first sentence of the Definition field from the CTD’s chemicals vocabulary
Chemical Pharmacodynamics: The Pharmacodynamics field of the DrugBank database (40)
Chemical General function: The General function field of the DrugBank database
Chemical Specific function: The Specific function field of the DrugBank database
Protein function: The function field of the UniProt database (41)
Embedded side information
In addition to the textual side information, we also evaluate entity embeddings trained via KBE methods, as they are capable of encoding topological information of KBs into dense vectors that can be used to infer relations between entities in the KB (42). For this, we experimented with multiple KBE methods trained on a graph representing the chemical–protein interactions in CTD (http://ctdbase.org/reports/CTD_chem_gene_ixns.csv.gz). We trained the models with the Deep Graph Library Knowledge Graph Embeddings library (43), optimizing the hyperparameters of embedding size |$\in \{200, 400, 600, 800, 1000\}$|, batch size |$\in \{128, 256\}$| and number of random negative samples |$\in \{50, 100, 200\}$| on a development split of the KB. Given an example with chemical c and protein p, we concatenate the corresponding KBEs ec and ep and feed them through a two-layer Multilayer Perceptron: |$h_e = \texttt{Dropout}(W_2 \texttt{Dropout}(\texttt{ReLU}(W_1 (e_c \circ e_p)))),$| where ReLU is a rectified linear unit (44) and Dropout a dropout layer (45) with probability 0.5. The resulting embedding he is then concatenated with the sentence embedding right before the output layer.
Apart from the KBEs we also investigate the incorporation of the contexts in which the chemical and protein entities are mentioned in the literature, as this can give further guidance about their connections to other biomedical concepts. For this purpose, we make use of the dense semantic entity representations provided by (46) which are learned in an unsupervised fashion using a language modeling task based on the complete PubMed corpus. The integration of these entity embeddings is analogous to that of the KBEs. In summary, we experimented with the following entity embedding methods:
We did not observe improvement with any of the entity embedding methods, thus we did not include them in our final model.
Additional training data through back-translation
We experimented with back-translating the DrugProt training data to introduce more textual variability. For this, we translate the training instances to German and French using pretrained machine translation models and then translate the result back into English and add it to our training data. We create translations with Facebook’s English-to-German transformer-based model trained on the Wmt news corpus (50) (https://huggingface.co/facebook/wmt19-en-de) as well as the English-to-German and English-to-French models by (51) (https://huggingface.co/Helsinki-NLP/) which were trained on the Opus corpus. Back-translations are generated by using the reverse variants of the respective models. We only use back-translated sentences in which we can find all mentioned entities of the original sentence by exact string matching and add them to the training set, others are discarded. In summary, we experimented with the following sets of back-translation models:
Opus and Wmt (+80,263 sentences)
Wmt (+26,507 sentences)
Note that we did not observe any improvement with back-translated data and thus do not use these data for training our final model.
Results
We evaluate the proposed model in two different settings. First, we evaluate the usefulness of the investigated modifications on the development set, individually optimizing the hyperparameters for each modification. Second, we submitted a selection of five different configurations to the official shared task evaluation on the hidden test set. For each of these two scenarios, we first describe the evaluation protocol and then the results. All reported scores are micro-averaged scores computed with the official DrugProt evaluation library (https://github.com/tonifuc3m/drugprot-evaluation-library).
Evaluation on DrugProt Development Set
We use the DrugProt development set to evaluate the modifications that we proposed above. We use a RoBERTa-large (52) as the baseline, initialized with the RoBERTa-large-pm-M3-Voc (https://github.com/facebookresearch/bio-lm) weights provided by (39). Then, for each modification, we search the best combination (on dev) of learning rate, number of epochs and batch size by performing an exhaustive grid search over the ranges described above using a fixed random seed (42). After finding the best hyperparameter configuration for the fixed random seed, we evaluate four more random seeds using the same hyperparameter configuration. When including also preliminary experiments, this leads to a total of 2647 training runs logged in the used experiment logging system (https://wandb.ai/). In some cases, a model fails to converge for a given random seed, so we drop the two seeds with the lowest F1 values and report mean and standard deviation of the remaining three, which leaves only converging models for all configurations but one. Furthermore, we evaluate an ensemble of these three runs that were also used to compute mean and standard deviation. We produce the ensemble prediction by averaging the predicted probabilities of each ensemble member. In preliminary experiments, we investigated ensembling models that were initialized with different pretrained language models, but found that ensembling only models derived from a single language model performed better on the DrugProt development set.
Results for this experiment can be found in Table 2. The only modification that achieves a higher F1 score than the |$78.6\%$| of the baseline is the addition of chemical definitions derived from CTD which leads to an F1 score of |$78.9\%$|. Ensembling three models leads to an improvement in F1 for all modifications except entity masking with an average gain of 1.8 percentage points (pp) in F1. All other modifications led to lower F1 scores than that of the baseline, for both single model and the ensembles. The lowest F1 score of 53% F1 was obtained when including entity embeddings computed with Rescal, because in this case only two of the five models converged and thus one run with a recall of 2.4% was included, which produced predictions for only a small fraction of the sentences. When using different pretrained transformers, results ranged from 75.6% F1 for BioMed RoBERTa to 78.6% for RoBERTa-large-pm-M3-Voc in the baseline.
Results of different model configurations on DrugProt development set. All scores are in percentage. Single results are mean and standard deviation of the best three runs across five different random seeds. Ensemble denotes results of an ensemble of the three best runs per configuration.
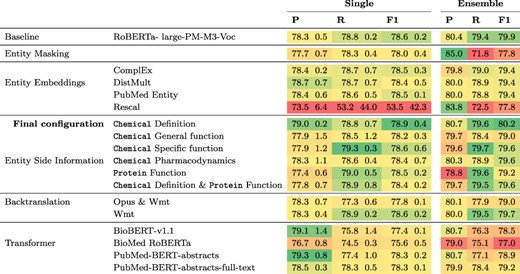 |
Evaluation on DrugProt Test Set
The DrugProt shared task allowed participants to submit a maximum of five runs for evaluation on the hidden test set. We selected the model configurations for these runs so that they could corroborate our findings on the development set. To achieve this, we prepared a run using our baseline and the two modifications that led to increased performance in our development set experiments: ensembling and entity descriptions. We slightly modified the configuration from the development set runs by increasing the number of ensemble members from three to ten and by adding the development data to our training set. We used the remaining four runs to systematically ablate the modifications as follows:
Run 1 (full configuration): Ensemble of 10 RoBERTa-large-PM-M3-Voc models with chemical definitions derived from CTD trained on training and development sets
Run 2 (single model): Single RoBERTa-large-PM-M3-Voc model with chemical definitions derived from CTD trained on training and development sets
Run 3 (no side information): Ensemble of 10 RoBERTa-large-PM-M3-Voc models trained on training and development sets
Run 4 (single model and no side information): Single RoBERTa-large-PM-M3-Voc model trained on training and development sets
Run 5 (no training on development set): Ensemble of 10 RoBERTa-large-PM-M3-Voc models with chemical definitions derived from CTD trained on the training set
The test set results can be found in Table 3. We observe the largest gain in performance of 1.7 pp F1 when adding chemical definitions and ensembling. Ensembling 10 models, only differing in the seed of the fine-tuning step, without chemical descriptions increases the F1 score by 1.4 pp, whereas adding chemical descriptions to a single model leads to a gain of 0.8 pp. Increasing the number of training examples by including the development set improves the F1 score by 0.2 pp.
Top: Results of the five submitted runs on the hidden test set of DrugProt. Bottom: Detailed results per relation type of Run 1. All scores are in percentage.
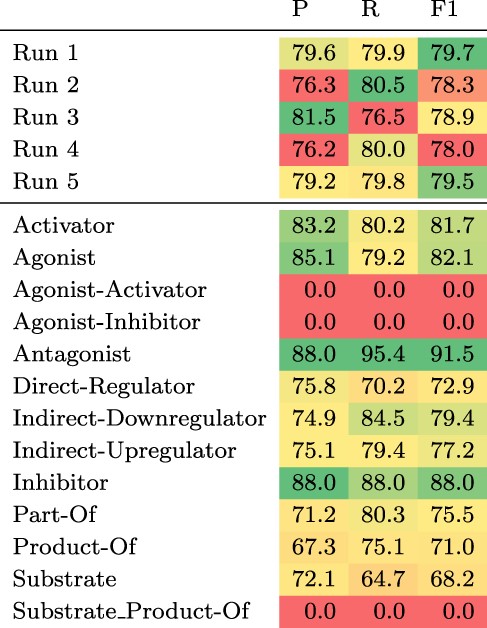 |
Considering the detailed results of our best submission (Run 1) for each relation type (see Table 3 bottom), there is a strong variability across different relation types with two types having an F1 score of zero (Agonist-Inhibitor and Substrate_Product-of), while the maximum F1 score is above 91% (Antagonist). The F1 scores correlate moderately with the number of training instances per relation type (Pearson’s R of 0.56). Both types with an F1 score of zero have very few training examples (13 and 24). However, for the other types there seem to be additional factors influencing performance. For instance, the ‘Substrate’ relation type has 2003 training examples, but the model achieves an F1 score of only 68.2%.
Discussion
Careful hyperparameter optimization is important for robust results
Our experiments on the development set suggest that baseline models can be surprisingly strong when tuned properly. We found that the most critical component to tune is the base language model, as replacing BioMed RoBERTa with RoBERTa-large-PM-M3-Voc led to an improvement of over 3 pp F1. We also analyzed the variability of the F1 scores when keeping the transformer fixed. For this, we looked at the lowest and the highest F1 scores for each transformer evaluated in the Transformer rows in Table 2. Here, F1 scores range from 66.3% to 78% for BioBERT-v1.1, 64.2% to 76.2% for BioMed RoBERTa, 0% (it failed to converge) to 78.4% for PubMedBERT-abstracts and from 65.8% to 78.5% for PubMed-BERT-abstracts-full-text. This indicates that the careful optimization of hyperparameters is crucial to optimize performance of pretrained transformers. We analyzed hyperparameter importance for these four pretrained language models with the functional analysis of variance (fANOVA) framework (53), which trains a random forest to predict the F1 score given the hyperparameter configuration and then uses the fANOVA framework to quantify hyperparameter importances. The results of this analysis can be found in Figure 2. Across all models, the most important hyperparameter to tune is the learning rate. For the other three hyperparameters, the ranking varies between models, with the difference between the average importances across models being negligible. Interestingly, the chosen random seed is as important as epochs and batch size when averaged across models, which suggests that this hyperparameter should also be routinely tuned for optimal performance. Moreover, these findings emphasize the importance of performing hyperparameter tuning for each model configuration. If neglected, this may lead to spurious findings of improvements under some modifications that are simply due to the high intra-configuration variability.
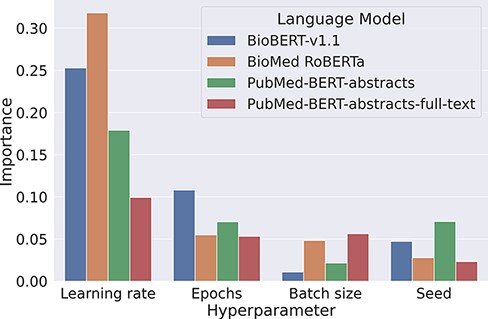
Overview about the importance of different hyperparameters using the fANOVA framework (53).
Knowledge Base Population Evaluation
A common use case for relation extraction models is KBP, in which the model is used to extract relations from a large collection of texts (7, 22). In addition to the shared task evaluation, we evaluate our model in such a KBP scenario, in order to gauge whether it could be used to assist KB curators. For this, we select the subset of four relations from the Therapeutic Target Database (TTD) (54) which are shared by the DrugProt corpus: activator, agonist, antagonist and inhibitor. Then, for each pair in this subset of TTD, we use PubTator Central (55), to collect all sentences from PubMed abstracts or PubMed Central full texts in which the pair co-occurs, discarding all pairs for which we do not find any sentence. Statistics for the resulting data set can be found in Table 4. To evaluate a model configuration, we use the respective model trained on the DrugProt training data to predict labels for each sentence using 0.5 as threshold. Finally, we aggregate over all sentences for a given pair by outputting all labels that were predicted for at least one sentence. We evaluate the models’ capability to assign the correct relation types to the TTD pairs by calculating precision, recall and F1 for the relation prediction. Note, that this might introduce a bias for the precision values, because we do not have access to negative samples.
Results of the KBP evaluation on the TTD data set. The results at the top are the ablation study, while the results at the bottom are the detailed results of the best performing model (baseline + ensembling). All scores are in percentage.
| P | R | F1 | # examples | |
|---|---|---|---|---|
| Baseline | 48.2 | 88.9 | 62.5 | – |
| + ensembling | 50.3 | 88.7 | 64.2 | – |
| + chemical definitions | 47.9 | 89 | 62.3 | – |
| + chemical definitions ensembling | 48.7 | 88.8 | 62.9 | – |
| Activator | 11 | 90.7 | 19.6 | 118 |
| Agonist | 49.7 | 88.4 | 63.6 | 667 |
| Antagonist | 42.1 | 89.1 | 57.1 | 660 |
| Inhibitor | 65.7 | 88.6 | 75.4 | 2437 |
The results for this evaluation can be found in Table 4. In terms of F1, we observe consistent gains through ensembling, both with and without chemical definitions (+0.6 pp F1/+1.7 pp F1). The addition of chemical definitions diminishes results in the single model setting as well as for ensembling (−0.2 pp F1/−1.5 pp F1). In addition to our results on the development set, this casts further doubts on whether chemical definitions are helpful. When inspecting the results of the best performing model (ensemble without chemical definitions) for each relation type individually, it becomes clear that the differences in F1 are almost exclusively due to variance in precision. The recall is consistently high for all examined relation types, ranging from 88.6% to 90.7%, whereas the lowest precision score is 11% and the highest is 65.7%.
We analyze the sources of errors by manually examining a random sample of 30 chemical-protein pairs for which the model extracted at least one false-negative or false-positive (FP) relation. We find that out of the 16 observed false-negative relations, 13 were because no sentence in our corpus allowed inference of the relation, 2 false negatives would have been possible to predict the context provided in the input sentence was insufficient and 1 required combining multiple pieces of information given in the sentence. For the 24 FPs, 22 are correct extractions which are not annotated in the TTD KB. Twelve of these correct extractions are due to unclear boundaries between the relation types of antagonist and inhibitor or of agonist and activator, where our model correctly extracts both relation types but only one is annotated in TTD. Of the two incorrect FPs, one is because of an incorrect gene normalization in PubTator Central and the other one is because the sentences from which the relation was extracted express it for a different gene than the annotated one. This suggests that we significantly underestimate the precision of the model, but a larger evaluation effort is required to confirm this.
Overall, we conclude from the results of our KBP evaluation that ensembles of properly tuned transformers achieve high accuracy for chemical–protein extraction ‘in the wild’ and might be helpful in KB curation efforts.
Are entity side information beneficial?
The results of our experiments on the DrugProt development set show strong performance gains for properly tuned baseline models. This applies equally for the ensembling of multiple models (see Tables 2 and 3) and also in the TTD evaluation setup (see Table 4). In contrast, the results for entity definitions are more mixed, we observe marginal gains on Drugprot’s development set and test set, but modest to larger drops in performance in the KBP evaluation.
To gain more insights about the differences when using entity definitions, we analyzed the prediction overlap of the baseline model and the model using chemical definitions.
Figure 3 highlights the overlap of ensembles of the two model variants regarding true positives (TPs) and FPs on the development set. In total, 2984 of the 3761 gold standard relations are identified by at least one of the two models. The overlap in TP of the model variants is very high (97.55%) and the number of relations found exclusively by only one model is almost symmetrical, differing only in 10 instances (63 vs. 73). The highest overlaps are observed in the relation types ANTAGONIST and SUBSTRATE, in which 196 of 198 (99.0%), respectively, 383 of 388 (98.71%) relations detected by both models match. Concerning FP predictions, the picture is a bit more diverse. Here, the predictions of both models only exhibit an overlap of 76.3%. The most marked differences can be recognized concerning the classes INDIRECT-DOWNREGULATOR and INDIRECT-UPREGULATOR, where the extended model only predicts 2 FPs each compared to 14 each of the baseline model. However, except for the two classes there is no clear pattern with regard to the distribution of errors across the different relation types. Analogous to the TPs the differences of both models in absolute terms are small and it is not clear whether this would also hold on a larger data set.
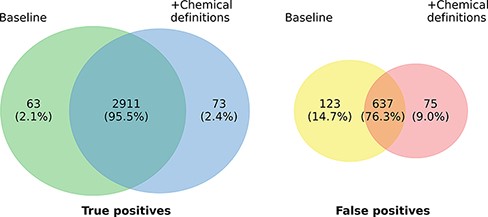
Prediction overlap concerning TPs (left) and FPs (right) between an ensemble of baseline models and an ensemble of models extended with chemical descriptions.
We also tried to identify patterns in cases where the extended model yields better predictions through manual analysis, but could not discern any clear underlying properties of sentences. We conclude that the improvements through the addition of chemical definitions need to be confirmed in further analysis and larger studies, which we leave for future work.
Comparison with competitors
We compared our approach to the three best other submissions to the shared task. Table 5 highlights the results of the teams on the hidden test set of DrugProt. All approaches are based on large pretrained BERT-based language models and utilize ensembles of multiple model instances for their best submission. The F1 scores achieved range from 77.6 to 79.7.
Results of the four highest ranked teams on the hidden test set of the BioCreative VII DrugProt shared task
| Team | P | R | F1 |
|---|---|---|---|
| Humboldt (our submission) | 79.6 | 79.9 | 79.7 |
| National Library of Medicine - | 78.5 | 80.5 | 79.5 |
| National Center for Biotechnology | |||
| Information | |||
| KU-AZ | 79.7 | 78.2 | 78.9 |
| University of Texas Health Science | 80.4 | 75.0 | 77.6 |
| Center |
The second best team (56) models the task in two different frameworks: (i) multi-class classification and (ii) sequence labeling. For the latter, given a candidate drug (protein) entity the goal of the model is to identify and label all corresponding protein (drug) entities which are involved in a relation with the candidate. Their best performing submission consists of an ensemble of multiple PubMed-BERT-based models of both frameworks using majority voting reaching an F1 score of 79.5.
Analogous to our approach, team KU-AZ (57) formulates the task as sentence-level classification problem. The authors investigate a distant supervision approach to extend the available training data. For this, the authors first train a model on the official DrugProt data set and then use it to automatically identify drug–protein relations in PubMed abstracts that are referenced in the CTD database. To reduce noise in the predicted relations they only keep relation pairs that are listed in the CTD database resulting in an data set with over 875K sentences. Using the additional data for model pretraining, however, shows slight drops in performance. Their best performing model configuration is based on an ensemble of 10 RoBERTa-large-PM-M3-Voc-based models learned on mixed splits of the DrugProt train and development.
Likewise, the fourth best team (58) models the task as sentence-level classification problem using different BERT flavors: PubMed-BERT, BioBERT, BioM-BERT and BioM-ALBERT (59). In contrast, to our model they perform entity masking for encoding the input entity pair under investigation. Their best models is based on an ensemble of 50 models trained on different splits.
Based on the description of the approaches it is hard to elicit all the technical details of the methods making the identification of a single reason for the (rather small) performance differences difficult. However, it is remarkable that only one of the other teams use the RoBERTa-large-PM-M3-Voc that shows a 0.3 pp higher F1 score over other BERT flavors in our experiments. Interestingly, team KU-AZ did not achieve any performance improvements through distantly supervised data confirming our observation that BERT-based baselines cannot be easily improved by additional data. In addition, all teams achieve performance improvements through model ensembling.
Conclusion
We described our contribution to the BioCreative VII DrugProt shared task, for which we developed a chemical–protein relation extraction model based on a relation classification framework and pretrained transformers. We performed an extensive search across hyperparameters and model configurations, which revealed that the choice of pretrained language model and ensembling had the largest impact on shared task performance. Furthermore, we found that including textual chemical definitions leads to small improvement on the DrugProt development and test sets but to diminished results in our KBP evaluation. The resulting model achieved an F1 score of 79.73% on the hidden DrugProt test set and was the first ranking submission of the 107 submitted runs in the official evaluation. We also evaluated the proposed model in a KBP setting on a distantly supervised chemical–protein relation extraction data set, which we created for this purpose. In this evaluation, we found that performance varied strongly with the relation type, suggesting that the model might be useful for KBP at least for some relations.
Acknowledgement
L.W. acknowledges the support of the Helmholtz Einstein International Berlin Research School in Data Science (HEIBRiDS). S.G. is supported by the Deutsche Forschungsgemeinschaft as part of the research unit ‘Beyond the Exome’. C.A. is supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany’s Excellence Strategy—EXC 2002/1 ‘Science of Intelligence’—project number 390 523 135.
Conflict of interest
There is no competing interest.
Author contributions statement
L.W., M.S., S.G., F.B., C.A. and U.L. conceived the experiments. L.W., M.S., S.G. and F.B. conducted the experiments. L.W., M.S. and C.A. analyzed the results. L.W., M.S., S.G., F.B., C.A. and U.L. wrote and reviewed the manuscript.

{kind=link}
{kind=link}
{kind=link}