





Abstract
Sepsis, one of the major challenges in the intensive care unit, is characterized by complex host immune status. Improved understandings of the phenotypic changes of immune cells during sepsis and the driving molecular mechanisms are critical to the elucidation of sepsis pathogenesis. Single-cell RNA sequencing (scRNA-seq), which interprets transcriptome at a single-cell resolution, serves as a useful tool to uncover disease-related gene expression signatures of different cell populations in various diseases. It has also been applied to studies on sepsis immunopathological mechanisms. Due to the fact that most sepsis-related studies utilizing scRNA-seq have very small sample sizes and there is a lack of an scRNA-seq database for sepsis, we developed Sepsis Single-cell Whole Gene Expression Database Website (SC2sepsis) (http://www.rjh-sc2sepsis.com/), integrating scRNA-seq datasets of human peripheral blood mononuclear cells from 45 septic patients and 26 healthy controls, with a total amount of 232 226 cells. SC2sepsis is a comprehensive resource database with two major features: (i) retrieval of 1988 differentially expressed genes between pathological and healthy conditions and (ii) automatic cell-type annotation, which is expected to facilitate researchers to gain more insights into the immune dysregulation of sepsis.
Introduction
Sepsis, defined as life-threatening organ dysfunction caused by a dysregulated host response to infection, is the leading cause of death in critically ill patients (1). Sepsis mortality remains high, up to 40% in septic shock patients (2, 3). And the long-term prognosis of septic patients also remains a serious issue. The 1-year mortality rate remains considerably high in sepsis survivors, with increased risks of readmission as well as cardiovascular and cerebrovascular complications (4, 5). Over the years, supportive treatment strategies for sepsis, including antibiotic administration, fluid resuscitation and organ function support, have been optimized. However, specific and effective therapeutics for sepsis remain unavailable (6, 7).
Immune dysfunction underlies the pathogenesis of sepsis (8). The feasibility of immunomodulation as a treatment for sepsis has been examined in a number of clinical trials, including early trials focused on inflammation blockade in the initial stage of sepsis and later trials focused on immunostimulatory agents. Some of those attempts showed efficacy in the improvement of certain aspects, such as organ function or disease severity score, or improved survival in a small subset of patients. However, none of them had demonstrated improvement in survival when applied to all septic patients (9–11). This is largely attributed to the complex, dynamic and heterogeneous immune status of septic patients and calls for a comprehensive and in-depth understanding of the immunopathology of sepsis (6, 12, 13). Therefore, elucidating the specific changes in phenotypes and signaling pathways of various immune and non-immune cell populations that constitute the defense system against invading pathogens is a critical question to address.
Loss of coordination of immune cell activities largely results in ineffective pathogen elimination and excessive tissue damage and can be detected at a very early stage, as shown by previous studies (14). Multiple mechanisms underlie the dysregulation of immune cell behaviors, including epigenetic regulation and metabolic reprogramming (15, 16). To uncover the molecular and genetic basis of the phenotypic alterations of immune cells, transcriptomic profiles need to be interpreted at the single-cell level, which requires the application of single-cell RNA sequencing (scRNA-seq). scRNA-seq allows transcriptomic profiling of tissues and organs at single-cell resolution, which has been applied to identifying disease-specific gene expression patterns through case-control studies and to detecting differences between cell populations (17, 18). Through scRNA-seq, cell-type specific expression patterns have been explored in Alzheimer’s disease, chronic myeloid leukemia, type 2 diabetes and other diseases (19–21). With the expansion of scRNA-seq data of human diseases, some scRNA-seq databases have been developed, such as CellMarker, PanglaoDB, scRNASeqDB, SCPortalen, SCDevDB and JingleBells (22–25). Common attributes of those databases include a query of gene expression levels across different datasets and clusters, differential expression analysis and retrieval of lists of marker genes for cell-type annotation. These databases provide researchers with a comprehensive view of the transcriptional patterns of individual cells under various pathological conditions, which is of great significance to the understanding of cellular heterogeneity, cell subpopulations and disease pathogenesis. However, at present, there is a lack of a single-cell transcriptomic database for sepsis. To address this issue, we developed Sepsis Single-cell Whole Gene Expression Database Website (SC2sepsis) (http://www.rjh-sc2sepsis.com/), a comprehensive database recording 1988 cell-type-specific differentially expressed genes (DEGs) and an automatic cell-type annotation tool dedicated for immune cells in septic condition.
Materials and methods
All data analysis processes were performed using R (R Project for Statistical Computing, RRID:SCR_001905) version 3.6.1 and the R package Seurat (SEURAT, RRID:SCR_007322) version 3.1.2.
Data preprocessing and cell-type annotation
ScRNA-seq data of each of the samples involved was deposited as a dataset in the public repositories. The 45 septic sample datasets and the 26 healthy control sample datasets were downloaded and merged into a diseased dataset and a control dataset, respectively. For basic quality control, cells with <200 nonzero genes detected and genes expressed in <3 cells were filtered out. Then, for each cell, all genes were ranked according to their expression levels in that cell. And two new matrices were generated by replacing the expression values in the expression matrices with rank numbers. For the original matrices, Z-score standardization was performed on the expression values.
The annotation results were manually inspected, and the ranges of cell types identified in both datasets were compatible with the major cell types occurring in human peripheral blood mononuclear cell (PBMC) samples. In addition, for users’ information, the marker genes (P < 0.05) of each cell type in the diseased dataset were generated through Seurat’s FindAllMarkers function with default parameters.
Differential expression analysis
To identify sepsis-related gene expression signatures of various cell types, after cell-type annotation, the diseased and control dataset underwent log normalization, batch effect correction through Seurat’s MultiCCA method. Then, DEGs (P < 0.05) of each cell type between the diseased dataset and the control dataset [e.g. between diseased natural killer (NK) cells and healthy control NK cells] were calculated using Seurat’s Wilcoxon rank-sum test with default parameters.
Automatic annotation of query datasets
SC2sepsis offers automatic cell-type annotation of matrix data obtained from septic patients uploaded by users. Specific scripts will enable the website to employ the correlation-based K-means for the K-nearest neighbors algorithm (see the section ‘Data preprocessing and cell-type annotation’) to annotate query datasets uploaded by users. And the diseased dataset derived from the four projects, which was completely annotated (see the section ‘Data preprocessing and cell type annotation’), will be used as the reference dataset.
To test the performance of the automatic annotation function of SC2sepsis, a 10 × 10-fold cross-validation (28) was performed. Each time a new dataset was generated by random sampling of the diseased dataset without replacement and was divided into 10 segments through stratified sampling: 9 of them were merged as the training dataset and the remaining 1 was regarded as the validation dataset. Stratified sampling was used to guarantee that the validation dataset contained as much cell types as the training dataset. Then, cell-type annotation of the validation dataset using the training dataset as the reference dataset was carried out. For every new dataset generated by random sampling, the process was performed for 10 times as each of the 10 segments was used as the validation dataset for one time. The average and the 95% confidence interval of the prediction accuracy with varying sample sizes of the random sampling are shown in Figure 1.
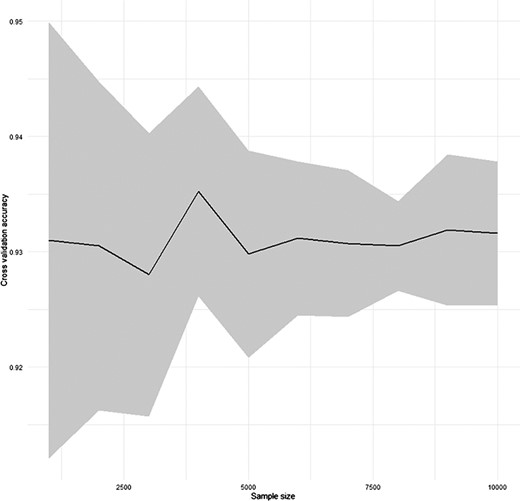
Result of the cross-validation for the performance of SC2sepsis automatic annotation function.
Results
Data collection and database content
We searched two public databases, the Gene Expression Omnibus (GEO) Database (https://www.ncbi.nlm.nih.gov/geo/) and the Single Cell Portal of Broad Institute (https://singlecell.broadinstitute.org/single_cell) for scRNA-seq studies on sepsis. ‘Human sepsis & single-cell RNA sequencing’ and ‘human sepsis & single-cell transcriptome’ were used as the keywords. Considering the fact that there are a limited number of scRNA-seq studies on sepsis, no additional filtering criteria were applied, and the search results were manually screened for eligible research projects that (1) enrolled patients meeting the Sepsis-2 or Sepsis-3 criteria of sepsis with or without healthy control populations (2); clearly specified the sample source and preparation processes (3) and have made their raw sequencing data or count matrices available for download. As a result, three projects from the GEO database and one project from the Single Cell Portal of Broad Institute (SCP) were included for further analysis. Those four projects comprise all the sepsis-related scRNA-seq studies conducted on human samples accessible in the GEO and SCP databases up to December 2021. All of them were conducted on PBMCs (SCP548 also included magnetic beads-enriched dendritic cells) isolated from patients and controls with their detailed information, as demonstrated in Table 1.
Information of the four datasets analyzed for the database construction
| Accession ID | Yeara | Sample size | Sampling time | Cell type | Number of cells from septic/healthy samples after quality control |
|---|---|---|---|---|---|
| GSE175453 | 2021 | Four septic patients and five healthy controls | Post-sepsis day 14–21 | PBMCs | 19 664/21 123 |
| GSE167363 | 2021 | Five septic shock patients and two healthy controls | 0 and 6 h from sepsis recognition | PBMCs | 38 214/14 647 |
| GSE151263 | 2020 | Seven septic patients (four with sepsis only and three with both sepsis and adult respiratory distress syndrome) | Within 24 h of initiation of mechanical ventilation | PBMCs | 16 021/0 |
| SCP548 | 2020 | Twenty-nine septic patients and 19 healthy controlsb | For the urinary tract infection (UTI) patients: within 12 h of the presentation to the emergency department; for the other patients: 24 h after hospital presentation and intravenous antibiotics administration | PBMCs, magnetic beads-enriched dendritic cells | 49 547/73 010 |
The year when the project datasets became public on the GEO database or the Single Cell Portal of Broad Institute.
SCP548 involved 19 healthy controls and subjects from 3 previous disease cohorts (referred to as cohorts a, b and c here): (i) 29 septic patients: 17 UTI patients with organ dysfunction from cohort a, 4 hospitalized septic patients from cohort b and 8 septic patients admitted to the medical intensive care unit (ICU) from cohort c; (ii) 19 healthy controls (iii) 17 non-septic patients (which were not included in our analysis): 10 patients with UTI but no organ dysfunction from cohort a and 7 non-septic ICU patients from cohort c.
Data of the four projects were reorganized into a diseased dataset and a control dataset according to the septic/healthy control status annotated in the original projects. After data preprocessing, cell-type annotation was performed on the two datasets independently, and the cell types identified are summarized in Supplementary File 1. Based on these data handling processes, SC2sepsis features two major functionalities (1): search and browse of the DEGs per cell type between the septic and healthy conditions (i.e. DEGs between each cell type in the disease dataset and the same cell type in the control dataset) (2); automatic cell-type annotation of septic datasets uploaded by users. The complete workflow of SC2sepsis is summarized in Figure 2.
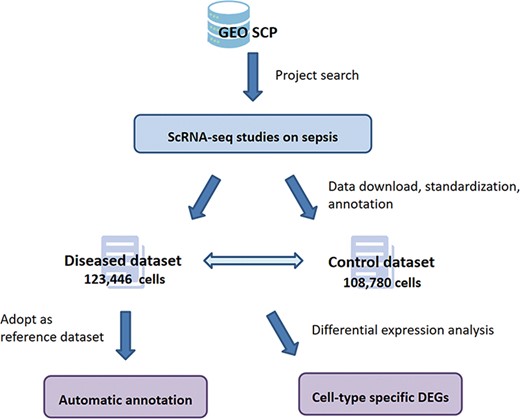
Overview of the workflow of SC2sepsis.
User interface
DEG search
SC2sepsis provides a search engine for retrieval of DEGs per cell type between the septic and healthy conditions. The search engine is linked to a standard gene library derived from the National Center for Biotechnology Information database (ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/GENE_INFO) to facilitate users to input the official names of the genes they would like to search for. As demonstrated in Figure 3A, once a character is typed into the panel, a drop-down menu of DEGs recorded in the database will appear accordingly for users to select the genes to be retrieved. The retrieval content feedback contains the following information (1): the cell types with significant differences in the expression level of the gene between the septic and healthy status and the corresponding log2 fold-change values (septic versus healthy) (2); the expression-level distribution of the gene across the septic and healthy control cells of each of the four projects.
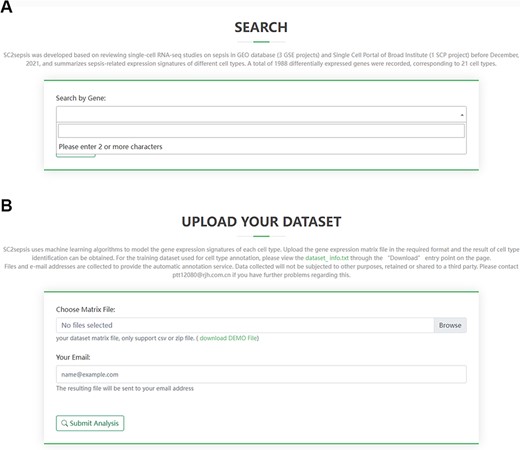
Overview of the search page for DEGs and the analysis page for users’ matrix data. (A) Search term input page. (B) Upload page for users’ matrix files.
For example, if the input is ‘lactate dehydrogenase isoform B (LDHB)’, the retrieval result will be displayed in two figures. The first one is a histogram (Figure 4A). Each column represents a cell type and indicates that the transcriptional level of LDHB is significantly different in macrophages, NK cells, CD34− pre-B cells, CD34+ pro-B cells, platelets and erythroblasts in septic patients compared to healthy controls. And the horizontal axis represents the log2 fold-change values of LDHB expression in the septic cells versus healthy cells. The second figure is a violin plot (Figure 4B). Each violin represents all the septic or healthy cells from a project, and the horizontal axis is the standardized expression value. The expression-level distribution of LDHB in each single-cell sequencing project can be visualized (click the violin to observe the median, minimum, maximum, first quartile and third quartile values of the expression value).
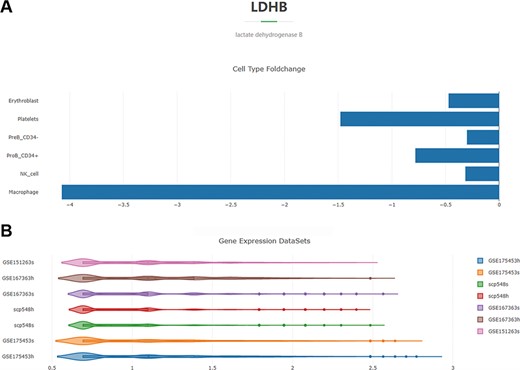
Overview of the DEG search result with the gene ‘LDHB’ as an example. (A) Histogram showing the log2 fold-change values of the cell-type-specific expression levels of LDHB in the septic versus healthy condition. (B) Violin plot of the expression-level distribution of LDHB across all the septic and healthy cells of the four projects.
Automatic annotation
Another major feature of SC2sepsis is automatic cell-type annotation of scRNA-seq data uploaded by users. As there have already been a number of tools and databases that facilitate the annotation of human cells under normal physiological conditions, SC2sepsis is dedicated to the annotation of data from patients with sepsis. SC2sepsis enables cell-type assignment to each cell independently based on the computation of its similarity in transcriptional signatures with each cell type in the diseased dataset, which contains disease-specific transcriptional signatures compared to HumanPrimaryCellAtlas or other commonly used reference datasets incorporated in SingleR. And the performance of the SC2sepsis annotation method was verified by cross-validation.
The analysis page for single-cell matrix data can be accessed through the ‘Home’ entry point on the front page of the database. Researchers can select matrix files or tab-separated values (TSV) files to upload and fill in the email address to receive the cell-type annotation results (Figure 3B). The annotation process takes ∼10 min and a demonstration of the result users will receive is provided in Supplementary File 2, where each row represents a single cell in the query dataset. For example, a typical row is sepsis1_GGGAATGCAGATAATG.5,0.568456758420006,0.557777416715459,“T_cells”. In this row, “sepsis1_GGGAATGCAGATAATG.5” is the barcode of the cell; “0.568456758420006” and “0.557777416715459” are the two correlation values computed in the last round of the fine tuning; “T_cells” is the cell type assigned to the single cell by the automatic annotation process.
The marker gene lists of each cell type in the diseased dataset (Supplementary File 3) can be found on the ‘Download’ page, which can also serve as input transcriptional signature files for other computational tools users would like to use for the annotation of their data.
Discussion
According to what we know, SC2sepsis is the only single-cell transcriptomic database of sepsis to date. On the basis of data integration from previous studies of small sample sizes and the adoption of a unified analysis algorithm to improve comparability across multiple datasets, it provides researchers with a comprehensive resource of DEGs between the septic and healthy status of 20 cell populations identified from human PBMC samples. These DEGs can be subjected to bioinformatic processes including clustering, Gene Ontology term enrichment, Kyoto Encyclopedia of Genes and Genomes pathway enrichment and protein–protein interaction) network analysis to provide users with clues to the interrelationships between these DEGs, as well as the biological processes and signaling pathways impacted. This information combined with further experimental verification will facilitate researchers to investigate key mechanisms underlying phenotypic alterations of immune cells during sepsis and other infectious and inflammatory conditions.
SC2sepsis is also an automatic annotation tool for scRNA-seq data collected from septic individuals. It adopts a classical machine learning classification algorithm and a well-recognized computational method for single-cell annotation to annotate datasets uploaded by users. It takes into account the expression of >2000 variable genes, which were selected from the whole genome by the algorithm and are considered to be more representative of the transcriptional pattern of each cell type. This attribute makes SC2sepsis superior to annotation based on a limited number of canonical cell markers manually collected from the literature. Nowadays, many comprehensive databases for marker genes of various species, tissues and cell types like CellMarker and PanglaoDB have also been developed. However, those resources are mainly derived from healthy cells. So, in this respect, transcriptional signatures derived from diseased datasets may be better applied to the annotation of data collected from septic individuals. In addition, the fact that every cell will be annotated independently avoids the problem of normalization in later annotations of new test datasets, which may be of different sequencing depth from the reference dataset.
Despite the efforts we put into the collection of datasets, the unneglectable fact is that currently available scRNA-seq datasets of sepsis are still limited in number and size. The amount of patients and cell types involved may not fully represent the heterogeneity of the population with sepsis and the diversity of patient immune status. With the development and application of scRNA-seq technology, the database will continue to be enriched and expanded. The reference datasets incorporated in this database not only will grow in number but also acquire a hierarchical system of cell-type labels, which means to enable the identification of cell subtypes from the major cell types already contained in the database. The recognition of rare cell populations will also be feasible. In conclusion, SC2sepsis is an easy-to-use platform for the exploration of sepsis-related gene expression signatures derived from scRNA-seq, which will be actively maintained to help researchers to investigate the mechanisms of sepsis occurrence, progression and immune dysregulation.
Supplementary data
Supplementary data are available at Database Online.
Acknowledgements
We would like to acknowledge the academic groups that have generated the scRNA-seq datasets used in the construction of this database.
Funding
National Natural Science Foundation of China (81772040, 81772107); Major Clinical Research Project of Shanghai Hospital Development Center (SHDC2020CR1028B); Scientific and Technological Innovation Act Program of Science and Technology Commission of Shanghai Municipality (18411950900).
Conflict of interest
None declared.
References
Author notes
contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}