





Abstract
This work presents Fragment Graph DataBase (FGDB), a graph database of ligand fragments extracted and generated from the protein entries available in the Protein Data Bank (PDB). FGDB is meant to support and elicit campaigns of fragment-based drug design, by enabling users to query it in order to construct ad hoc, target-specific libraries. In this regard, the database features more than 17 000 fragments, typically small, highly soluble and chemically stable molecules expressed via their canonical Simplified Molecular Input Line Entry System (SMILES) representation. For these fragments, the database provides information related to their contact frequencies with the amino acids, the ligands they are contained in and the proteins the latter bind to. The graph database can be queried via standard web forms and textual searches by a number of identifiers (SMILES, ligand and protein PDB ids) as well as via graphical queries that can be performed against the graph itself, providing users with an intuitive and effective view upon the underlying biological entities. Further search mechanisms via advanced conjunctive/disjunctive/negated textual queries are also possible, in order to allow scientists to look for specific relationships and export their results for further studies. This work also presents two sample use cases where maternal embryonic leucine zipper kinase and mesotrypsin are used as a target, being proteins of high biomedical relevance for the development of cancer therapies.
Database URL: http://biochimica3.bio.uniroma3.it/fragments-web/
Introduction and related work
Fragment-based drug design (FBDD) is an efficient alternative to traditional high-throughput screening (HTS). It relies on the identification of small and often weakly potent bioactive molecules that can be later expanded or linked together to generate novel drug-like leads. The main differences between FBDD and HTS are the composition and size of the libraries and the assay methods used for hit identification. Indeed, due to the lower affinity of the fragments, FBDD screenings make use of more sensitive techniques, while HTS needs more specific screening to reduce the number of false positives identified (1). FBDD has become of increasing interest in both academia and industry due to many advantages over traditional HTS (2). Thanks to their physicochemical properties, fragments can be successful even for those targets for which HTS has failed (3). FBDD yields relatively more hits in comparison to HTS (4, 5), due to the inverse relationship between molecular complexity and binding probability (6). Moreover, a reduced chemical complexity results in a broader exploration of the potential binding site of a target protein, thanks to the enhanced binding promiscuity provided by the fragments (7). A library of fragments covers a much larger chemical space than a similar-size collection of larger molecules. Small ligands boast an improved ligand efficiency compared to larger ligands, while lead compounds generated from fragment hits usually feature enhanced physicochemical properties (8). Therefore, the possibility to use a smaller chemical library to explore a wider chemical space made FBDD projects more accessible and affordable compared to HTS (9). This is proved by the discovery of Vemurafenib, the first fragment-derived drug, which employed just 6 years to move through the various clinical trial phases and to be approved by the Food and Drug Administration (2). A crucial step in FBDD is the selection of the proper fragment library, to enhance the hit rate and reduce the number of false positives. A fragment is defined as a chemically stable, low-molecular-weight and highly soluble organic compound. Fragments can resemble chemical moieties and functional groups often found in drug-like compounds. They are often characterized by an affinity in the range of micromolar to millimolar. To define the desired biochemical properties of a fragment, Congreve and coworkers (10) drew an analogy with Lipinski’s rule of five and postulated the so-called ‘rule of three’. Following these guidelines, an ideal fragment must feature a molecular weight less than 300 Da with three or less hydrogen bond donors, three or less hydrogen bond acceptors and a CLogP no higher than 3. However, there is evidence that blindly adhering to ‘the rule of three’ for the design of a fragment library may lead to the exclusion of compounds that would otherwise show up as hits (11). Once the fragments library is defined, it is screened against the target biomolecule. The screening can be carried out with both biophysical and in silico techniques, including ligand-observed NMR, X-ray crystallography, isothermal titration calorimetry and molecular docking.
The contribution of this work consists of providing the scientific community with a graph database of ligand fragments, Fragment Graph DataBase (FGDB), along with all of the corresponding information associated with them, including their frequencies of contact with the amino acids, the ligands they are contained in and the proteins the latter bind to. FGDB has been designed and inherently built as a graph database consisting of nodes (entities) and directed edges (relationships). As a matter of fact, a graph database stores its data as a graph instead of tables (as in a Relational Database Management System or RDBMS), providing a greater expressive power with respect to a traditional RDBMS and allowing graph-based search and retrieval mechanisms. At the same time, the so-called Atomicity, Consistency, Isolation, and Durability (ACID) properties of an RDBMS are not guaranteed, as it is the case with No-SQL databases. Being built ‘offline’ and provided in a read-only fashion, FGDB is not negatively affected by this limitation but is instead empowered by its nature as a graph and can be effectively browsed in a variety of ways.
The paper is organized as follows. In the remainder of this section, related work is discussed by separating graph databases in biochemistry from fragment-based approaches. In Section 2, the graph database is described in greater detail, including the underlying data and how it was generated, along with its schema and its entities (nodes) and relationships (edges), as well as the details of its technical implementation. Section 3 provides an overview of the core features of the web application that enables users to access, browse, query and export the database and shows some sample use cases for biochemical scientists. Finally, Section 5 draws the conclusions.
Graph databases
Although data relationships can be easily represented by using a graph abstraction, the optimization of queries requires additional data structures. For this reason, Graph Database Management Systems (GDBMSs) are preferable (12); as they do not require further engineering to implement the usual traverse algorithms, graph databases have been proven successful in numerous biomedical applications, where typically the relational paradigm is not particularly efficient. The idea of GDBMSs is to decouple the most significant part of the computational requirements to specific infrastructure nodes, increasing the performance of the algorithms.
In biochemistry, for example, biochem4j (13) connects different data resources, such as NCBI, UniProt, MNXref and ChEBI. A typical query output shows a user-friendly interface (originated by Neo4j) that can be further explored (for example, by showing the different relationships between organisms, enzymes, reactions and chemicals) by the user, thanks to extended interactivity. The authors claim that biochem4j can assemble data from a wide set of resources generating a repository including data on 1 544 257 named organisms, 2 457 504 enzymes, 36 765 reactions and 256 230 chemical species. Queries are performed by exploiting the Cypher query language that, because of its complexity, should be manipulated by means of advanced graphical user interfaces.
GREG (The Gene Regulation Graph Database) (14) is an integrative database based on a web interface to provide the users an integrative analysis of transcriptional regulation. While the graph database connects different data sources (such as 4DGenopme, iRefIndex and many others): starting from the concept of bin (an arbitrary segment of a chromosome, which is more likely to be added to a graph with respect to a single base pair. Approaches using bins can easily organize heterogeneous information interaction, for example, the one from proteins and DNA), GREG provides an integration between protein and lncRNA data (i.e. biomolecules) and genomic coordinates (based on numerical intervals). Other features consist of the harmonization of bins and small protein-binding sites with data originated from chromatin interaction methods.
EpiGeNet (15) is a graph database for storage and querying data for molecular events, occurring at different stages of colorectal oncogenesis. The query process is articulated in (i) stage-specific molecular events, (ii) most frequently observed genetic and epigenetic interdependencies in colon adenoma and (iii) paths connecting key genes reported in CRC and associated events. EpiGeNet is able to visualize molecular events related to CRC initiation and progression.
BioGraph (16) is a web application querying a graph database called BioGraphDB. The authors motivate the application by presenting a study case concerning the analysis of microRNA in breast cancer. The novelty introduced by BioGraph consists of Gremlin, a popular open-source and vendor-agnostic graph computing framework used to query the database, whose access is possible, thanks to pre-defined templates and customized requests.
The Fragment Network (17) is a graph database used to search for a chemical space around a compound of interest: considering each compound articulated into rings, linkers and substituents, the chemical space can be regarded as a network. Specifically, the database consists of nodes and edges that are produced by removing iteratively these groups from the parent molecule. As the number of nodes in the database can be of the order of the hundreds of millions, the search algorithm needs to be efficient. The deployed methods consist of (i) a recursive building algorithm adding a compound into the Fragment Network, resulting in a set of nodes and edges described by a set of attributes, and (ii) a query algorithm taking as input a molecule in the fashion of non-isomeric canonical SMILES string. A start node is initialized to the matching node or, in case the search fails, the set of nodes and edges related to the query; a compound is produced and merged into the network. By default, the search query returns all available compounds from graph nodes whose distance (measured in edges) from the start node is zero, one, or two. Other methods, such as deletions, replacements and result sorting, are available.
Fragment-based approaches
GraphSim TK [see (18) and references therein] is a tool offering different fingerprint types to perform similarity measurements in two dimensions, ranging from Path to LINGO approaches. The user is offered the capability to highlight the size of the enumerated paths, the atom and the properties associated with the bond.
FragVLib (19) is a free software tool aiming at studying similarity measures across ligand−receptor complexes databases. The result provides important information exploited to identify binding pockets sharing similar characteristics to those describing a target receptor. The pocket similarity search process requires, as an initial pre-requisite, a target receptor and a database consisting of native ligand−receptor complexes. The algorithm is articulated into the following steps: (i) determination of the interfacial atoms (composed of the receptor and the ligand atoms included within a cutoff distance) by using a dedicated algorithm called almost-Delaunay tessellation, (ii) representation of the interfacial atoms by means of an un-directional graph, (iii) matching the interfacial graph of the target complex and the interfacial graph of each native complex in the database and (iv) copying the ligand nodes and edges directly connected for a found match, with the matched sub-graph into the target receptor.
CREDO (20) is a database of protein–ligand interactions, denoting contacts as structural interaction fingerprints. Among its most notable features, it is completely scriptable, thanks to an application programming interface. More specific features include (i) the realization of molecular shape descriptors with ultrafast shape recognition, (ii) fragmentation of ligands in the Protein Data Bank (PDB), (iii) sequence-to-structure mapping and, finally, (iv) the identification of approved drugs. However, CREDO potential can be largely improved by optimizing the methods creating the dataset and the way by which ligands are recognized, with attention to peptide ligands.
The PDB in Europe-Knowledge Base [PDBe-KB (21)] is a collaborative resource for structural and functional annotations of the structure data deposited in the PDB. Among its many features, PDBe-KB provides the PDBe Graph Database in which each PDB entry is the root of a tree connected to chains and entities, in turn connected to residues for which available annotations are provided. From the ‘Motifs and Sites’ section of the database, for each ligand, contact statistics with amino acids can be obtained. However, this information is apparently not available from the web resource at the level of ligand fragments.
Finally, in (22), the authors present DeepFrag, a deep convolutional neural network used to predict the correct fragments provided the structure of a receptor−ligand complex. The idea is to optimize the process recognizing the chemical fragments that might be added to a known ligand to enhance its binding affinity.
Materials and methods
Data content
By leveraging the method described in (23) and briefly summarized in the following subsection, the authors have generated and assembled a collection of more than 17 000 ligand fragments from the entries available in the PDB (24). These fragments are linked to the amino acids with which they are in contact with a given frequency, the ligands they are contained in and the proteins the latter bind to: as such, amino acids, ligands and proteins are also included in FGDB with their minimal subset of information and their hyperlinks to the corresponding entries of the PDB.
Fragment generation
The fragment-residue contact datasets were derived from LIBRA’s binding site database (whose last update date is on 28 April 2022) (25–27). LIBRA’s binding site is a semi-exhaustive binding site database derived from PDB (24) containing 163 473 binding sites. Each entry, identified by a unique code (defined as XXXX_YYY_Z where XXXX is the PDB ID, YYY the ligand ID and Z the chain letter), contains a ligand, its binding site (amino acids within 3.5 Å) and the surrounding environment (amino acids within 5 Å). A ligand’s neighbor residues are divided into binding residues and environments. A preprocessing step involving the removal of incorrectly formed entries was carried out, since a number of entries were either empty or only contained heteroatoms. Complexes whose structures were determined by NMR were not included in the dataset. In addition, to remove data redundancy and ensure data accuracy and the biological relevance of the contacts identified, three different filtering procedures were carried out.
In order to better underline the contact between a ligand’s fragment and the binding site’s amino acids, only proteins with high-quality crystal structures were included. This was achieved by filtering out complexes solved with a resolution higher than 2.5 Å. To ensure the biological significance of the identified contacts, non-biologically relevant compounds such as crystallization additives were removed from the datasets; this filtering procedure was performed according to a blacklist of compounds derived from BioLip (28) (https://zhanggroup.org/BioLiP/ligand_list). In addition, entries containing carbohydrates, oligopeptides, ions and nucleic acids, not relevant for this study, were manually removed. Lastly, to avoid bias due to over-represented proteins in the dataset, a sequence clustering procedure was performed using CD-HIT (29) with an identity threshold of 40%. Two complexes were kept even when their sequence identity was above the threshold if they sufficiently bound different ligands. In order to do this, among each protein cluster, the ligands were grouped based on their similarity. The similarity was measured through the Jaccard distance calculated on their circular Morgan fingerprints (30) and stored on a distance matrix. Based on this matrix, a Taylor-Butina clustering algorithm (31) was applied, and the protein complex containing the centroid of each ligand cluster was added to the final database. For the ligand clustering phase, a Jaccard distance threshold of 0.3 was employed. When an entry contained multiple equivalent binding sites for the same ligand, only one binding site was considered, adding the information concerning additional residues present in the other binding sites.
The contact identification procedure involved the fragmentation of all the dataset’s ligands. For each binding site, the ligand was fragmented using the BRICS fragmentation algorithm (32). The data manipulated through a Python script were stored in a dictionary containing the code of the binding site as the key and the set of fragments generated from the fragmentation as the values. Then, each binding site residue was depicted as an ensemble of tagged points (depicting non-hydrogen atoms, with the tag representing the origin residue). A KD-Tree algorithm was employed for the spatial searching phase. Starting from the coordinates of each fragment, a residue was considered a neighbor of the fragment if there was at least one tagged point belonging to it, within a distance of 4.5 Å from a fragment’s atom. Contacts were registered in 20-bit arrays, with each bit representing a different amino acid. For each fragment, the contact array was stored in a dedicated dictionary using the fragment’s SMILES as the key. The occurrences of the contact between a given fragment and each amino acid were obtained by summing up contact arrays sharing the same SMILES.
Schema of the graph database: entities and relationships
The entities with the corresponding relationships making up the database schema are the following:
Aminoacid. It represents a building block of a protein. The 20 canonical amino acids are considered and stored in terms of their three-letter code.
Fragment. This is the core entity of the graph database, representing a fragment of a ligand uniquely identified by a SMILES, as produced via the technique described in Section 2.2. Each Fragment is assigned a progressive alphanumeric id in the form ‘FRAGX’, where X is the progressive number, and a picture showing its molecular structure. A Fragment is involved as a subject of the following relationships:
CONTACTS: a Fragment may contact an Aminoacid with a certain frequency. This relationship (or directed edge) expresses such a contact, having a Fragment as its subject (or start node) and an Aminoacid as its object (or end node); the relationship is weighted by the number of times a Fragment contacts an Aminoacid.
IS_CONTAINED_IN: a Fragment is contained in one or more Ligands, and different Ligands may contain the same fragments. This relationship (or directed edge) expresses this containment, having a Fragment as its subject (or start node) and a Ligand as its object (or end node).
Ligand. A small molecule from the PDB, stored with its alphanumeric three-character code and the corresponding link to the PDB. A Ligand is involved in the following relationship:
BINDS_TO: a Ligand can bind to one or more Proteins: this is expressed via such a relationship, having a Ligand as its subject (start node) and a Protein as its object (end node).
Protein. An entry from the PDB, stored with its alphanumeric four-character code and corresponding link to the PDB. A Protein is only involved as the object (end node) of the BINDS_TO relationship mentioned above.
Populating the graph database
The information related to the ligand fragments, namely their contact frequencies with the 20 amino acids, the ligands they are contained in and the proteins the latter bind to, were stored in three textual files. Each of the first two files featured one row per fragment and included the contact frequencies with the 20 amino acids for each fragment and the list of ligands they are contained in, respectively. The third file mapped the ligands with the proteins they bind to, with a row for each of such mappings.
As such, the graph database has been populated by parsing these files and generating the corresponding entities (nodes) and relationships (edges) accordingly. Technical details on the actual programming language and libraries used in this regard are reported in the next subsection.
Implementation and technical details
The mechanism for generating the ligand fragments has been implemented in Python v. 3.6+ and takes advantage of the following libraries: RDKit v.2020.09.1 and its implementation of the BRICS algorithm.
The underlying framework of the graph database is Neo4j v. 3.5.18, whose query language is Cypher. The procedure for populating it as well as the back-end application for manipulating it and querying it has been developed in the Java language, v. 1.8+, and relies on Spring Data Neo4j repositories and APIs. These APIs are able to reduce the number of Cypher queries and corresponding methods to be manually written.
The web application has been developed in Java and JavaScript and deployed on an Apache Tomcat application server v. 9+. The front-end graphical library that enables the visualization of the graph database and the graphical queries is Popoto.js.
Results and discussion
Core features and graphical queries
The access to FGDB is provided via an online web application, where the data stored in the graph database can be browsed and queried in a number of fashions: these include standard, form-based queries and graphical queries. Furthermore, data can also be exported in CSV format for later re-use, offline experiments and interoperability with external applications. All of these features are further detailed in the next paragraphs.
Browsing the database
The graph database can be browsed by accessing the homepage of the web application (http://biochimica3.bio.uniroma3.it/fragments-web/). The initial view shows the nodes corresponding to the types of entities stored and relationships, along with the details of a subset of Fragments. Figure 1 shows the landing page and describes its elements in greater detail. The numbers shown on the bottom right of each node correspond to the number of entities of such a type featured in the database.
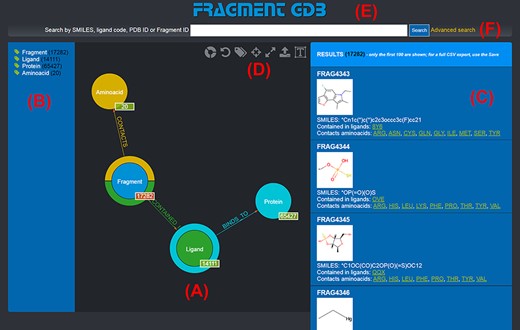
Landing page of the FGDB. (A) The main panel of the graph database, where the entities and their relationships are shown and which allows users to browse the data and perform graphical queries for filtering them out. (B) The left side taxonomical panel shows the types and number of entities stored in FGDB; it is immutable regardless of the queries performed and allows users to individually filter a specific type of entity to be visualized. (C) The right side panel shows a subset (up to 100) of the Fragments resulting from the currently executed query, displaying the number of the total results and providing, for each entity shown, its corresponding information in terms of identifier, molecular structure, SMILES, list of ligands where the Fragment is contained and list of amino acids contacted; the latter two are hyperlinked with the corresponding entry of the PDB. (D) Upper toolbar of the main panel, providing users with a number of useful functionalities, from left to right: hiding/showing clickable relationships around the entities (the colored crowns/half-crowns around the Fragment and Ligand nodes), resetting the graph to the last executed query, hiding/showing the left taxonomical panel, centering the graph, making the graph full-screen, exporting the current graph and enlarging/shrinking the size of the nodes’ textual labels. (E) The basic search field where users may search for Fragments by SMILES of the Fragment, ligand code of the contained ligands, PDB ID of the proteins the contained ligands bind to or Fragment identifier. (F) The hyperlink to the advanced search panel, where users may carry out more complex queries against the database.
This view is dynamic: users may interact with it in a number of ways, from clicking and dragging nodes for a clearer view, to exploring the actual entries for a given entity type by single-clicking an entity node, up to applying graphical filters to produce a specific result (more on that in Section 3.1.3).
By clicking on a node, it is possible to browse a number of the entities of the given type: for instance, by clicking on the ‘Aminoacid’ node, a subset of up to 10 entities of this type are shown as nodes expanding from the entity type node; to browse for more entities of such a type, it is possible to use the left/right arrows appearing on the entity type node. This is shown in Figure 2. Obviously, this mechanism is suitable for entities whose total number is limited. Should a user want to search for specific entities, a variety of query mechanisms are of course available and are discussed in the next subsections.
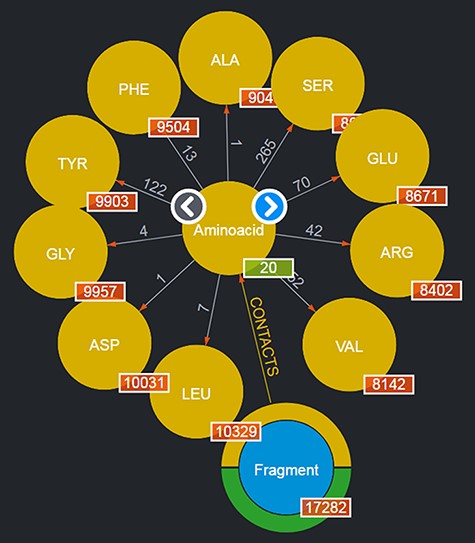
Entities of type ‘Aminoacids’ shown after clicking on the corresponding entity type node. The view shows up to 10 entities and allows users to browse for more by using the left/right arrows in the center. The numbers displayed on the edges, in this case, correspond to the total number of Fragments featured in the DB contacting the given amino acid.
The upper toolbar of the main panel where the graph is displayed also provides a number of useful functionalities, from customizing or resetting the current view up to exporting the entities shown (more details on that in the caption of Figure 1 and in Section 3.1.5).
Basic queries
Searching for specific information in the FGDB can be done with a variety of query mechanisms The most basic of these mechanisms lies in filling out the text field placed at the uppermost part of the page. There, users can look for Fragments by typing either a SMILES, a three- or two- character ligand code, a four-character PDB ID or, if known, an internal Fragment ID of the database.
For instance, by looking for a given ligand, e.g. MC3 (1,2-dimyristoyl-rac-glycero-3-phosphocholine), it is possible to obtain as a result a graph showing the number of Fragments contained in such a ligand, the number of Proteins the ligand binds to, etc., as depicted in Figure 3. The returned graph features the entity found in place of the given entity type node, and by clicking on the entity type nodes connected to it, it is possible to browse the returned entities with the same mechanism described in Section 3.1.1. If no entity is found with the given user query, the page is simply reloaded and the standard graph is shown.
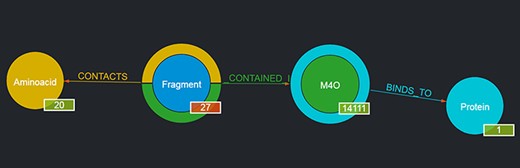
Output of a basic query where the user had looked for ‘M4O’. The results page shows the searched ligand instead of the generic ligand entity type in the graph, along with the number of Fragments that ligand contains (in this case, 27) and the number of proteins it binds to (in this case, 1); the 27 Fragments contact as a whole all of the 20 amino acids; thus, 20 is displayed as their number. By clicking on the corresponding entity type node (e.g. Fragment), it is possible to browse each of the entities returned and in the case of Fragments to see their details on the rightmost panel as well (not shown in the figure).
Graphical queries
Another option a user has for querying FGDB is to directly interact in a graphical way with the schema nodes shown in the main panel. In fact, the graph shown is an interactive representation of the underlying database schema and allows for the construction of graphical queries by using the mouse.
As depicted in Figure 1, the default view of the graph shows four schema nodes, each corresponding to an entity type of FGDB (Fragment, Aminoacid, Ligand and Protein). Each node corresponding to an entity type that features outward relationships (namely, all of them with the exception of Proteins) has a circular colored crown or half-crown around it, with the same color scheme as the nodes the outward relationships connect with. For instance, the Fragment node sports a yellow half-crown, corresponding to the ‘CONTACTS’ relationships with the yellow-colored Aminoacid node, and a green half-crown, corresponding to the ‘IS_CONTAINED_IN’ relationship with the green-colored Ligand node, etc.
Both the entity type nodes, the colored crowns and the relationships, are interactive in order to ‘build’ a graphical query. Specifically:
Filtering by specific entities. By left-clicking on a node, such a node gets expanded showing the actual entities of that type, grouped in clusters of 10 elements that can be browsed with the left or right arrows provided, as also discussed in Section 3.1.1; obviously, this browsing has limited usefulness for looking for a specific entity when no other restrictions or filters are applied. By left-clicking on a specific entity around the entity type node, the latter gets replaced by the code of the specific entity, thus applying a filter on the graph that gets changed accordingly. For instance, by clicking on the Aminoacid node, the first 10 amino acids are brought up, and by clicking on one of them (e.g. GLY), the resulting graph gets filtered accordingly, showing only those Fragments contacting GLY and their other related entities (Ligands in which they are contained along with the Proteins they bind to). Right-clicking on a node where a filter has been applied resets the filter.
Conjunctive, disjunctive and negated queries. By left-clicking on a crown or half-crown, a relationship of that type is added to the graph, enabling users to add additional conjunctive filter conditions on specific entities (i.e. each relationship is connected in AND with one another). For instance, after selecting GLY as in the earlier example, a user can click on the yellow half-crown that will consequently sprout another ‘CONTACTS’ relationship with another Aminoacid node; by selecting TYR in that node, the resulting query (and thus filter) would mean: ‘give me all of the Fragments (and related Ligands and Proteins) that contact both GLY and TYR’.
This mechanism can also be used to construct disjunctive queries. In order to do this, after clicking on GLY, for instance, and selecting it as the specific entity on the Aminoacid node, if a user left-clicks again on it and then selects another amino acid (e.g. ALA), the entity node will then show ‘GLY or ALA’ and will thus produce a query retrieving all of the Fragments that contact either GLY or ALA.
A relationship/edge can be also deleted by hovering the mouse over them and clicking on the ‘X’ symbol that appears upon the relationship’s name, thus removing that particular filter condition.
Finally, by holding CTRL down and then clicking on an entity type node, it is possible to negate the given filter and thus produce negated queries: for instance, by left-clicking (while holding down the CTRL button) on the earlier selected TYR, the resulting query would be: ‘give me all of the Fragments that are not in contact with TYR (plus any other additional filter selected, of course)’. If the user left-clicks on an entity type node without first selecting a specific entity for it, the whole relationship connecting it with the closest entity type is negated (for example, if ‘Aminoacid’ is negated, no Fragments are returned, since there is no ligand fragment that does not contact at least one amino acid) (For Mac OSX users, at the time of writing of this manuscript, this feature is not available, due to a current issue with the Popoto.js library used for displaying and querying the underlying graph database, which apparently prevents any browser running on Mac OSX (Chrome, Safari, etc.) from correctly detecting the pressing of the CTRL button. The authors are sorry for any inconvenience this might cause. It is thus advisable, for Mac OSX users, to resort to the advanced query panel instead in order to perform negated queries).
Needless to say, the user can mix and match conjunctive, disjunctive and negated queries as they see fit. An example of a graphical query is shown in Figure 4.
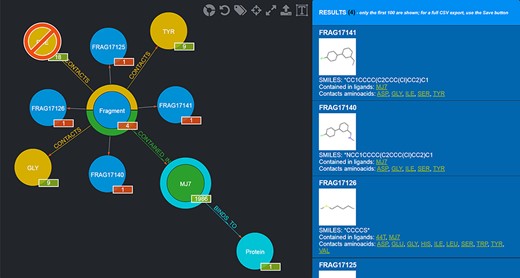
Example of a graphical query. The query performed shows those Fragments that contact both TYR and GLY but not PHE and are contained in MJ7. This view was thus produced by filtering Fragments as follows: firstly, by clicking on the Aminoacid node and selecting TYR by clicking on it; secondly, by clicking on the yellow half-crown around the Fragment node for bringing up another ‘CONTACTS’ relationship and doing the same selection as before for GLY; thirdly, by repeating the previous step, but this time using CTRL + click to select PHE, in order to negate the relationship; fourthly, clicking on the Ligand node and selecting MJ7; finally, by clicking on the Fragment node (whose amount was reduced to 4, i.e. the only Fragments fulfilling the selected conditions) and displaying the actual Fragments resulting from the query; their details are shown in the right-side panel. The numbers on the bottom right of the nodes have the following semantics: the numbers in the red background refer to the resulting Fragments (4 total, each Fragment showing 1 as they are indeed individual); the other numbers refer to the Aminoacids they contact as a whole (9), the total number of Ligands that Fragments contacting the chosen Aminoacids are contained in (1986) and the number of proteins the selected ligand (MJ7) binds to (1).
Advanced queries
Should a user need a more expressive power in querying the graph database, FGDB also provides an advanced search panel, reachable from the corresponding link on the right of the main search text field. This panel enables users to specify more complex queries and thus filters on the database entries, in terms of a composition of conjunctive, disjunctive and negated queries. Specifically, it is possible to fill out a number of variable rows expressing a subject-relationship-object triple (node-edge-node), each corresponding to a given query to be concatenated in AND with the others, which are made up of three elements: the first element is a drop-down list for selecting the subject node entity, the second is a drop-down list for selecting the relationship (or its negated version) and the third is a text field where a specific entity can be used as input, or a sequence of entities separated by semicolons in which all of the entities are connected in OR with one another. Figure 5 shows the advanced search panel filled out with some sample values for performing a more complex query, while Figure 6 displays the actual results of such a query.
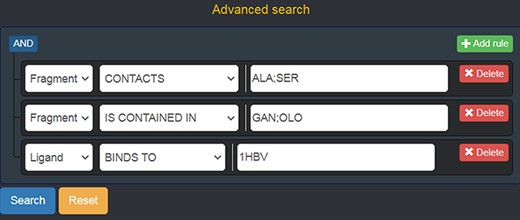
Advanced search panel, with some text fields populated as an example. The semantics of the query shown here is the following: “retrieve all of the Fragments which contact the amino acids ALA or SER, AND which are contained in the ligands with code GAN or OLO, AND whose containing Ligands bind to the protein identified by the PDB ID 1 HBV.
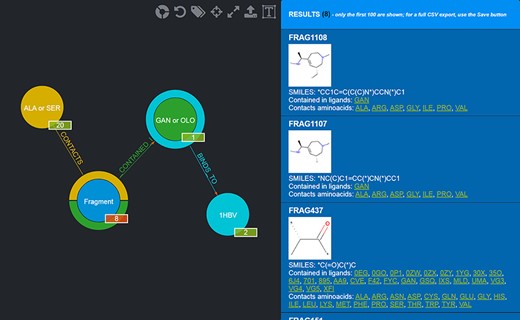
Results of the advanced query shown in Figure 5. Twenty-eight Fragments are returned (whose details are provided in the rightmost panel).
Data export
At any given time, the graph displayed with the corresponding entities, either derived from a basic, graphical or advanced query, or showing the whole content of the Fragment GDB, can be exported in CSV format by clicking on the corresponding export icon on the upper right of the main panel (the second one from the right, with the arrow pointing upwards). When dealing with a huge number of search results, for specific use cases (as in Section 3.2, for instance) or for checking out the entire database, the export mechanism allows users to overcome the obvious display limits of the web pages. The resulting CSV files are conveniently provided as a standard representation for offline analyses and for integration with a user’s specific workflows.
Sample use cases
To illustrate possible uses of the database in the context of drug design, maternal embryonic leucine zipper kinase (MELK), a promising target for anticancer therapies is analyzed as use case. MELK is a member of serine-threonine kinase family that has been shown to take part in the regulation of fundamental cellular processes such as proliferation and apoptosis. Overexpression of this protein is associated with poor prognosis in a variety of cancer types, and therefore, MELK is the target of many drug design efforts in the search for potent and specific inhibitors (33).
As can be seen in Figure 7, the MELK active site can be divided into two subpockets (34) in which a number of inhibitors are observed to bind. The first subpocket is lined by residues Ile17, Leu27, Ala38 and Tyr88, while the second is lined by Lys40, Glu57, Asn137 and Asp150.
![Schematic representation of the MELK in complex with the inhibitor 3-[2-(phenylcarbamoyl)-5-(1H-pyrazol-4-yl)phenoxy]propan-1-aminium developed by a FBDD approach. The red ellipses indicate the two subpockets of the active site in which inhibitors bind [PDB code: 4D2T (34)].](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/database/2022/10.1093_database_baac044/1/m_baac044f7.jpeg?Expires=1787771506&Signature=GNG1AoCKsaqagCJ-Lt0J9CFZ~YBxyIGETz0KhH-g6h5h1A82Zw5NvSAhPkrMJ7P2Y3ZuJ2ELE-ZPkfkofKSNOt08vGFAwVEnu0ghO97IgVTtpPAMTnSOsO0Bz8JA7YOPZ-nwonTQb9~otxJV~dvlzrLke15Hxuz9rig7z8RkHEDSaQHt4P5tnSjUBi4BZ32oxhweMAE~5s5MFuw-XvQ5O2GrxbhF7Lp-JkxlXhhuKyD3P7oS2-2B6KTiZF0uKgG-ojpSZoM9hoyv9z1K86I-nPGua0nO0MK951KvFw4ngLKW~Vtv64ZeLTqhIUXXOKoXXXvPW6dE3w7ov83Em2BSZA__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Schematic representation of the MELK in complex with the inhibitor 3-[2-(phenylcarbamoyl)-5-(1H-pyrazol-4-yl)phenoxy]propan-1-aminium developed by a FBDD approach. The red ellipses indicate the two subpockets of the active site in which inhibitors bind [PDB code: 4D2T (34)].
One possible use of the database is the selection of fragments known to bind in a specific protein microenvironment. From this viewpoint, two different queries can be done to select from the database fragments observed to contact amino acids characterizing, respectively, the first and the second subpocket (Figure 8) and use the two sets of fragments for an FBDD approach. Fragment information, including their SMILES, can then be exported in the form of CSV files.
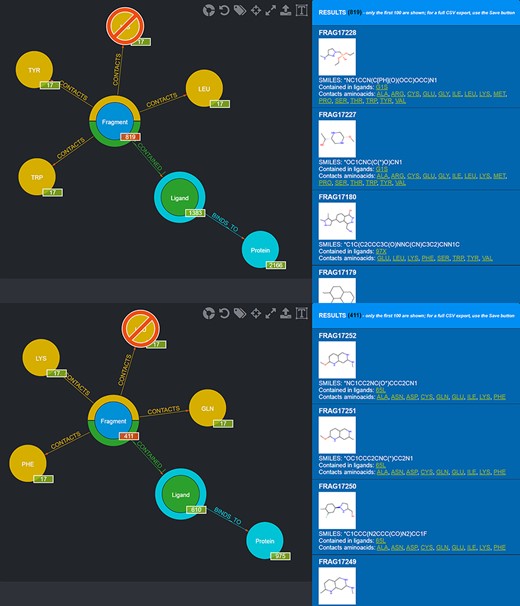
Screenshot of the graphical queries done to extract from the database those fragments observed to contact the amino acids lining the first (top panel) and the second (bottom panel) subpocket of MELK with, on the right side, a preview of the results of the queries.
Alternatively, through a textual query using all the PDB codes of MELK structures solved in complex with small molecule inhibitors, all the fragments building up these inhibitors can be retrieved from the database exported and used for a combinatorial drug development approach.
Another use case is the exploitation of FGDB for the selection of fragments for the development of specific inhibitors targeting members of protein families characterized by high structure and sequence similarity. One such family is that of serine proteases that represents a significant challenge for the development of selective inhibitors (35). As an example, the oncogenic protease mesotrypsin and the closely related kallikrein-6 have been selected. Figure 9 displays a superimposition of the three-dimensional structures of these two proteases showing the high structural and sequence similarity. FGDB can be used to select fragments that are likely to bind the desired target, i.e. mesotrypsin, while minimizing the probability of off-target effects, in this case, the binding of fragments to the related protease kallikrein-6. As it can be seen in the figure, also mesotrypsin’s active site can be divided into two subpockets, one lined by Tyr94, Leu99 and Trp215 and the other by Phe41, Lys60 and Gln192. Kallikrein-6 displays a very similar environment but with two notable exceptions, namely the substitution of Leu99 by a His residue in the first pocket and the substitution of Phe41 by a Leu residue in the second pocket.
![Schematic representation of the three-dimensional structure of the oncogenic protease mesotrypsin [light brown, PDB ID 3L33 (36)] and the closely related kallikrein-6 [blue, PDB ID 1L2E (37)] highlighting similarities and differences in the two subpockets adjacent to the catalytic triad (unlabeled Ser, His and Asp residues) on opposite sides.](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/database/2022/10.1093_database_baac044/1/m_baac044f9.jpeg?Expires=1787771506&Signature=O4~J5YYeb-bfyl4fEF8~HriCZHSnmnwEoAv9Wg6IKDJGUTmMgBLcnlBMnsZdEQdEM9od2rA8wkx61uY89eWrmqo1vFxVJfi73egMzE3opdABLA4SzeuxE04c883BYl5FabPvO2HfQnu6lgb2qEjPABPhH83tIORG-1Ndz8lZ3ysgutnQTlmtK6CiNDQflAEUVRnVkvsJrbazEHZZxuWR8wcboyDDk2Z9gPIAEs2dGMkgA7J~OKG32u2iirXjh8LEXemGBG~a-IL8sJ2ZHlGhrEahyQrTDZw8q62tsD6AA2JsK1QfdnT~4I21VCWBeGZZxqmj2Z5nXNyXWqzq71r5yw__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Schematic representation of the three-dimensional structure of the oncogenic protease mesotrypsin [light brown, PDB ID 3L33 (36)] and the closely related kallikrein-6 [blue, PDB ID 1L2E (37)] highlighting similarities and differences in the two subpockets adjacent to the catalytic triad (unlabeled Ser, His and Asp residues) on opposite sides.
Also in this case, two different queries can be carried out to select those fragments that contact amino acids characterizing the first and the second subpocket of mesotrypsin, respectively, while excluding fragments observed to contact the different amino acids found in the kallikrein-6 subpockets (Figure 10).
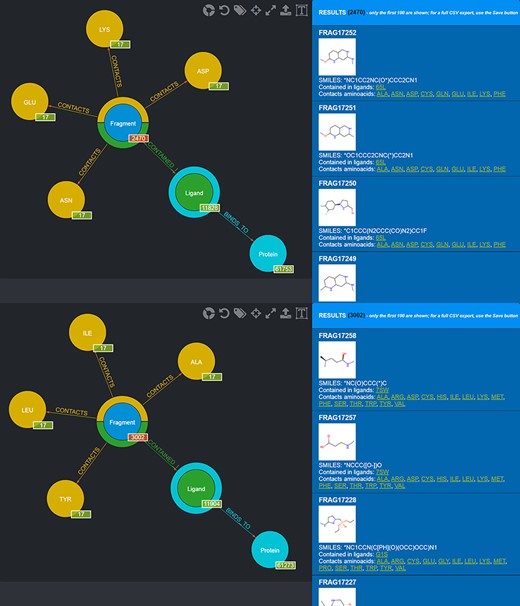
Screenshot of the graphical queries done to extract from the database those fragments discussed in the mesotrypsin use case, with, on the right side, a preview of the results of the queries.
The CSV files corresponding to the different queries carried out for the above-mentioned use cases (MELK’s subpocket 1, MELK’s subpocket 2, total fragments obtained from co-crystallized ligands in MELK structures, mesotrypsin subpocket 1 and mesotrypsin subpocket 2) are available for download at the following link: http://www.computationalbiology.it/software/fgdb/export_fragments_CSV_files.zip.
Conclusion
In this work, FGDB, a graph database of ligand fragments, has been presented. FGDB includes more than 17 000 ligand fragments with their related info in terms of SMILES representation, their frequencies of contact with the 20 amino acids, the ligands which they are contained within and the proteins the latter bind to. FGDB is freely accessible via a web application that allows users to conveniently browse and query the database in a variety of ways, in accordance with personal preference and scientific usefulness: from basic textual searches, to interactive graphical queries, up to advanced filters. Data resulting from queries or from the whole database can be exported in the standard CSV format at any given time. FGDB can be a useful and effective support for building target-specific libraries within the context of computational drug design campaigns based on ligand fragments. In this regard, this work has also provided two use cases related to two target proteins, MELK and mesotrypsin, highly relevant for developing cancer therapies.
Supplementary data
Supplementary data are available at Database Online.
Funding
This work was supported by the Italian Ministry of University and Research, under grants ‘Dipartimenti di Eccellenza’ (L. 232/2016, Art. 1, c. 314–337) and PRIN (Grant No. 2017483NH8) to F.P.
Conflict of interest
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}