





Abstract
Posttranslational modifications (PTMs) refer to amino acid side chain modification in some proteins after their biosynthesis. There are more than 400 different types of PTMs affecting many aspects of protein functions. Such modifications happen as crucial molecular regulatory mechanisms to regulate diverse cellular processes. These processes have a significant impact on the structure and function of proteins. Disruption in PTMs can lead to the dysfunction of vital biological processes and hence to various diseases. High-throughput experimental methods for discovery of PTMs are very laborious and time-consuming. Therefore, there is an urgent need for computational methods and powerful tools to predict PTMs. There are vast amounts of PTMs data, which are publicly accessible through many online databases. In this survey, we comprehensively reviewed the major online databases and related tools. The current challenges of computational methods were reviewed in detail as well.
Introduction
Posttranslational modifications (PTMs) are covalent processing events that change the properties of a protein by proteolytic cleavage and adding a modifying group, such as acetyl, phosphoryl, glycosyl and methyl, to one or more amino acids (1). PTMs play a key role innumerous biological processes by significantly affecting the structure and dynamics of proteins (2, 3). Generally, a PTM can be reversible or irreversible (4). The reversible reactions contain covalent modifications, and the irreversible ones, which proceed in one direction, include proteolytic modifications (5). PTMs occur in a single type of amino acid or multiple amino acids and lead to changes in the chemical properties of modified sites (6). PTMs usually are seen in the proteins with important structures/functions such as secretory proteins, membrane proteins and histones. These modifications affect a wide range of protein behaviors and characteristics, including enzyme function and assembly (7), protein lifespan, protein–protein interactions (8), cell–cell and cell–matrix interactions, molecular trafficking, receptor activation, protein solubility (9–14), protein folding (15) and protein localization (16). Therefore, these modifications are involved in various biological processes such as signal transduction, gene expression regulation, gene activation, DNA repair and cell cycle control (17–19). PTMs occur in various cellular organelles including the nucleus, cytoplasm, endoplasmic reticulum and Golgi apparatus (5).
Proximity ligation assay (PLA) is a novel immunoassay technology that can be used to study PTMs (20). In addition to PLA, immunoprecipitation (IP) is utilized in several different PTM detection assays (21). However, the combination of mass spectrometry with IP strategy is a more effective method (22). Nevertheless, large-scale detection of PTMs is very costly and challenging. In recent years, computational methods for predicting PTMs have attracted a considerable attention (5, 16, 17, 23–26).
The rest of this paper is structured as follows. In the section ‘The 10 most studied PTMs’, the 10 most studied PTMs will be described. Major PTM databases will be reviewed in the section ‘The 10 most studied PTMs’ as well. In the section ‘Involvement of PTMs in diseases and biological processes’, involvement of PTMs in diseases and biological processes will be discussed. Then, computational methods for predicting PTMs will be described in the section ‘Computational methods for predicting PTMs’. Finally, tools for PTM prediction will be reviewed in the section ‘Tools for PTM prediction’.
The 10 most studied PTMs
There are more than 400 different types of PTMs (27) affecting many aspects of protein functions. According to the dbPTM (6), one of the most comprehensive PTM databases, there are 24 major PTMs, with more than 80 experimentally verified reported modified sites. Figure 1 provides a visualized summary of the current major PTM data according to the dbPTM. According to Figure 1, we can see that some of these major PTMs occur more frequently and have much more been studied. Three main PTMs, based on the dbPTM database, are phosphorylation, acetylation and ubiquitination, which comprise more than 90% (∼827 000 sites out of ∼908 000) of all the reported PTMs Accordingly, each amino acid undergoes at least three different PTMs, and Lys undergoes the largest number of PTMs (15 PTM types). Moreover, based on the whole dbPTM data, Cys and Ser are also modified with at least 10 PTM types. Finally, one can see that phosphorylation on Ser is the most reported PTM type.
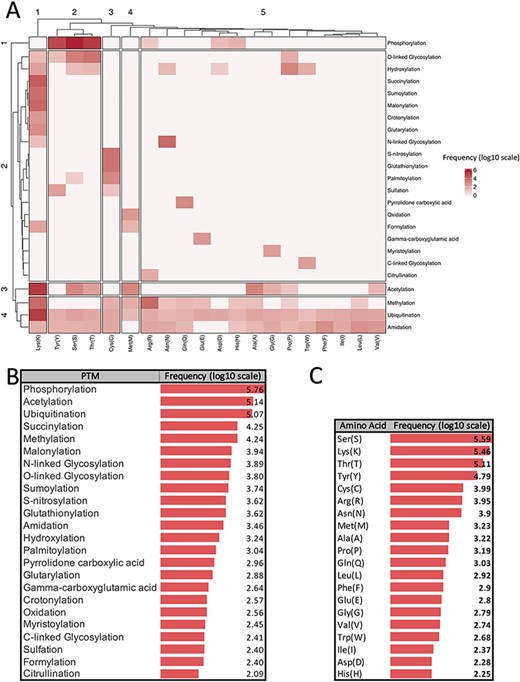
Summarized information of major PTMs (24 PTMs with more than 80 experimentally verified reported modified sites) according to the dbPTM databank (October 2020). All frequencies are shown in log scale. (A) Clustergram indicating the frequency of each PTM on different amino acids. (B) Frequency of major PTMs. (C) Frequency of each amino acid that was reported as a modified site.
Figure 1A shows a clustergram, indicating the division of the PTMs into four clusters as one can see each phosphorylation, and acetylation has been considered as a separate cluster due to their different patterns of modification on the amino acids. On the other hand, ubiquitination, methylation and amidation are the PTMs with many different target residues and have been clustered as a group. According to the clustergram, amino acids have been divided into five clusters. Amino acid Lys is the most different amino acid based on the PTM pattern.
Panels B and C in Figure 1 show the frequency of PTM types and amino acids in the dbPTM database in log scale, respectively. According to Figure 1, it is observed that phosphorylation, acetylation and ubiquitination are the most frequent PTMs.
Roughly speaking, according to the type of the modifications, these PTMs can be categorized into three main groups. First and second groups are those PTMs that include the addition of chemical and complex groups to the target residue, respectively. The first group and the second group include glycosylation, prenylation, myristoylation and palmitoylation. Those PTMs that contain addition of polypeptides to the target residue comprise the last group, and these PTMs are ubiquitylation and SUMOylation. Figure 2 shows a graphical timeline for the discovery of these major PTMs. In this timeline, the organisms in which each PTM was discovered for the first time also have been depicted. In the following subsections, the 10 most studied PTMs, out of these major ones, are described in more detail.
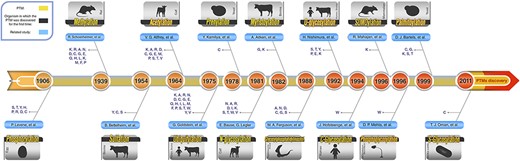
Schematic PTM discovery timeline for 10 major PTMs: phosphorylation (28), methylation (29), sulfation (30), acetylation (31), ubiquitylation (32), prenylation (33), myristoylation (34), SUMOylation (35), palmitoylation (36), different types of glycosylation (N-glycosylation (37), O-glycosylation (38), C-glycosylation (39) and S-glycosylation (40)), phosphoglycosylation (41) and glycosylphosphatidylinositol (GPI anchored) (42). For each PTM, target residue(s) and the organism in which the related PTM was discovered for the first time are shown.
Phosphorylation
Protein phosphorylation was first reported in 1906 by Phoebus Levene with the discovery of phosphate in the protein vitellin (phosvitin) (28). However, it took another 20 years before Eugene Kennedy described the first enzymatic phosphorylation of proteins (43). This process is an important reversible regulatory mechanism that plays a key role in the activities of many enzymes, membrane channels and many other proteins in prokaryotic and eukaryotic organisms (44, 45). Phosphorylation target sites are Ser, Thr, Tyr, His, Pro, Arg, Asp and Cys residues (6), but this modification mainly happens on Ser, Thr, Tyr and His residues (46). This PTM includes transferring a phosphate group from adenosine triphosphate to the receptor residues by kinase enzymes (Figure 3A). Conversely, dephosphorylating or removal of a phosphate group is an enzymatic reaction catalyzed by different phosphatases (47). Phosphorylation is the most studied PTM and one of the essential types of PTM, which often happens in cytosol or nucleus on the target proteins (48). This modification can change the function of proteins in a short time via one of the two principal ways: by allostery or by binding to interaction domains (49).
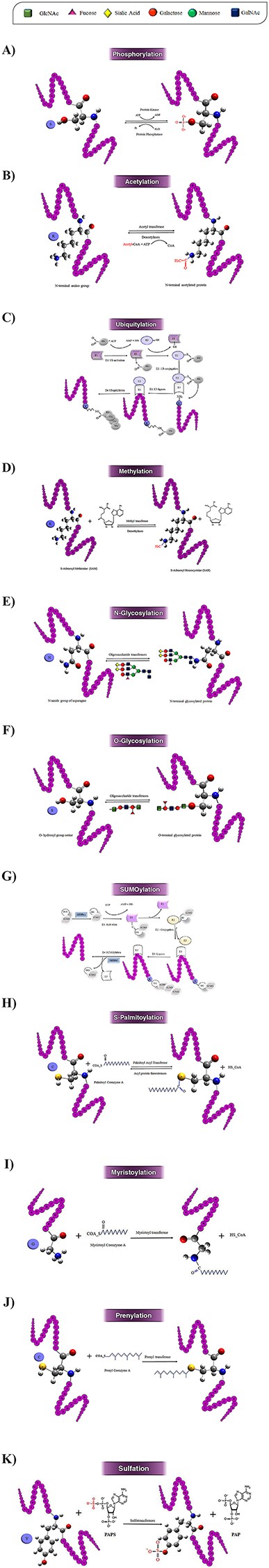
Schematic illustration of the 10 most studied PTMs including Phosphorylation (A), Acetylation (B), Ubiquitylation (C), Methylation (D), N-glycosylation (E), O-glycosylation (F), SUMOylation (G), S-palmitoylation (H), N-myristoylation (I), Prenylation (J), and Sulfation (k).
Phosphorylation has a vital role in significant cellular processes such as replication, transcription, environmental stress response, cell movement, cell metabolism, apoptosis and immunological responsiveness (12, 50, 51). It has been shown that disruption in the pathway of phosphorylation can lead to various diseases such as cancer, Alzheimer’s disease, Parkinson’s disease and heart disease (24, 52, 53).
Acetylation
The first acetylation modification in proteins was discovered by V.G. Allfrey in 1964 in isolated calf thymus nuclei in vitro (31). Acetylation is catalyzed via lysine acetyltransferase (KAT) and histone acetyltransferase (HAT) enzymes. Acetyltransferases use acetyl CoA as a cofactor for adding an acetyl group (COCH3) to the ε-amino group of lysine side chains, whereas deacetylases (HDACs) remove an acetyl group on lysine side chains (Figure 3B) (54). There are three forms of acetylation: Nα-acetylation, Nε-acetylation and O-acetylation. Nα-acetylation is an irreversible modification, and the other two types of acetylation are reversible (55). These three forms of acetylation occur on Lys, Ala, Arg, Asp, Cys, Gly, Glu, Met, Pro, Ser, Thr and Val residues with different frequencies (6), although the acetylation is more reported on Lysine residue. Nε-acetylation is more biologically significant compared to the other types of acetylation (55).
Acetylation has an essential role in biological processes such as chromatin stability, protein–protein interaction, cell cycle control, cell metabolism, nuclear transport and actin nucleation (56–58). According to the available evidence, acetylated lysine is vital for cell development, and its dysregulation would lead to serious diseases such as cancer, aging, immune disorders, neurological diseases (Huntington’s disease and Parkinson’s disease) and cardiovascular diseases (56, 59, 60, 61).
Ubiquitylation
Ubiquitylation is one of the most important reversible PTMs. This modification was firstly studied in 1975 by Gideon Goldstein (32). This modification is a versatile PTM and can occur on all 20 amino acids (Figure 2). However, it occurs on lysine more frequently. This PTM has a major role in the degradation of intracellular proteins via the ubiquitin (Ub)–proteasome pathway in all tissues (62). In ubiquitylation, a covalent bond befalls between the C-terminal of an active ubiquitin protein (a polypeptide of 76 amino acids) and Nε of a lysine residue of the protein (63). Ubiquitin can occur in mono- or poly-ubiquitination forms on substrate proteins through specific isopeptide bonds by receptors containing ubiquitin-binding domains. Ubiquitylation is catalyzed by an enzyme complex that contains ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin ligase (E3) enzymes (Figure 3C). Ubiquitinated proteins may be acetylated on Lys, or phosphorylated on Ser, Thr or Tyr residues, and lead to dramatically altering the signaling outcome (64). Ubiquitylation modification in substrate proteins can be removed by several specialized families of proteases called deubiquitinases (64).
Ubiquitination plays important roles in stem cell preservation and differentiation by regulation of the pluripotency (65). Ubiquitylation has also played a vital role in many various cell activities such as proliferation, regulation of transcription, DNA repair, replication, intracellular trafficking and virus budding, the control of signal transduction, degradation of the protein, innate immune signaling, autophagy and apoptosis (12, 66, 67). Dysfunction in the ubiquitin pathway can lead to diverse diseases such as different cancers, metabolic syndromes, inflammatory disorders, type 2 diabetes and neurodegenerative diseases (68–70).
Methylation
Research on methylation dates back to 1939 (29). Nonetheless, just recently, with the identification of new methyltransferases (such as protein arginine methyltransferases (PRMTs), and histone lysine methyltransferases (HKMTs)), has attracted more and more attention (71). Methylation is a reversible PTM, which often occurs in the cell nucleus and on the nuclear proteins such as histone proteins (1, 72). Methylation occurs on the Lys, Arg, Ala, Asn, Asp, Cys, Gly, Glu, Gln, His, Leu, Met, Phe and Pro residues in target proteins (6). However, lysine and arginine are the two main target residues in methylation, at least in eukaryotic cells (73, 74). One of the most biologically important roles of methylation is in histone modification. Histone proteins, after synthesis of their polypeptide chains, are methylated at Lys, Arg, His, Ala or Asn residues (75). Nε-lysine methylation is one of the most abundant histone modifications in eukaryotic chromatin, which includes transferring the methyl groups from S-adenosylmethionine to histone proteins via methyltransferase enzyme (Figure 3D). In eukaryotes, methylated arginine has been observed in histone and non-histone proteins (76).
Recent studies have shown that methylation is associated with fine tuning of various biological processes ranging from transcriptional regulation to epigenetic silencing via heterochromatin assembly (77). Defect in this modification can lead to various diseases such as cancer, mental retardation (Angelman syndrome), diabetes mellitus, lipofuscinosis and occlusive disease (12, 78, 79).
Glycosylation
One of the most complex PTMs in the cell is glycosylation, which is a reversible enzyme-directed reaction (12). Glycosylation occurs in multiple subcellular locations, such as endoplasmic reticulum, the Golgi apparatus, cytosol and the sarcolemma membrane (80). Glycosylation occurs in eukaryotic and prokaryotic membranes and secreted proteins, also nearly 50% of the plasma proteins are glycosylated (14). In this modification, oligosaccharide chains are linked to specific residues by covalent bond (see Figures 3E and F). This enzymatic process, which is catalyzed by a glycosyltransferase enzyme, usually occurs in the side chain of residues such as Trp, Ala, Arg, Asn, Asp, Ile, Lys, Ser, Thr, Val, Glu, Pro, Tyr, Cys and Gly (6); however, it occurs more frequently on Ser, Thr, Asn and Trp residues in proteins and lipoproteins (13). According to the target residues, glycosylation can be classified into six groups: N-glycosylation, O-glycosylation, C-glycosylation, S-glycosylation, phosphoglycosylation and glypiation (GPI-anchored) (5, 12). N-glycosylation and O-glycosylation are two major types of glycosylation and have important roles in the maintenance of protein conformation and activity (81).
Glycosylation has a great role in many important biological processes such as cell adhesion, cell–cell and cell–matrix interactions, molecular trafficking, receptor activation, protein solubility effects, protein folding and signal transduction, protein degradation, and protein intracellular trafficking and secretion (9–14). It has been shown that the defect in this process has a significant effect on the development of various diseases like cancer, liver cirrhosis, diabetes, HIV infection, Alzheimer’s disease and atherosclerosis (12, 14, 82).
SUMOylation
Small Ubiquitin-Related Modifier (SUMO) protein was primarily discovered in 1996 by Rohit Mahajan in the Ran GTPase-activating protein (RanGAP) (35). SUMOylation takes place via SUMO (83) that has a three-dimensional structure similar to ubiquitin protein and has been discovered in a wide range of eukaryotic organisms (84). SUMOylation can occur in both cytoplasm and nucleus on lysine residues (85). SUMO family has three isoforms in mammals, four isoforms in humans, two isoforms in yeasts and eight isoforms in plants (1). SUMOylation occurs as a modifier in ε-amino group of lysine residues in target protein through a multi-enzymatic cascade (86). In this reaction, SUMO is connected to a lysine residue in substrate protein by covalent linkage via three enzymes, namely activating (E1), conjugating (E2) and ligase (E3). Also, it is separated from the target protein by a specific enzyme protease—SUMO (Figure 3G) (87). Often, SUMOylation modifications occur at a consensus motif WKxE (where W represents Lys, Ile, Val or Phe and X any amino acid) (88).
SUMOylation plays a major role in many basic cellular processes like transcription control, chromatin organization, accumulation of macromolecules in cells, regulation of gene expression and signal transduction (89, 90). It is also necessary for the conservation of genome integrity (91). Also, there are many reports on major role of SUMOylation in development of a variety of human diseases including cancer, Alzheimer’s disease, Parkinson’s disease, viral infections, heart diseases and diabetes (83, 91–93).
Palmitoylation
An important class of PTMs, called lipidation, includes covalent attachment of lipids to proteins. The first report of the covalent modification of proteins with lipids dates back to 1951 (94). These PTMs are taken place via a great variety of lipids like octanoic acid, myristic acid, palmitic acid, palmitoleic acid, stearic acid, cholesterol, etc. Myristoylation, palmitoylation and prenylation can be considered as the three main types of these lipid modifications (95, 96). Palmitoylation is described in this subsection, and the other two important ones are described in the subsequent subsections.
Palmitoyltransferases (PATs) were first identified in yeast in 1999 by Doug J. Bartels (36). Palmitoylation is the covalent attachment of fatty acids, like palmitic acid on the Cys, Gly, Ser, Thr and Lys (6). S-palmitoylation contains a reversible covalent addition of a 16-carbon fatty acid chains, palmitate, to a cysteine via a thioester linkage (Figure 3H) (97). Palmitoyl-CoA (as the lipid substrate) is attached to the target protein by a PAT and removed via acyl protein thioesterases (98).
Mostly, S-palmitoylation occurs in eukaryotic cells and plays critical roles in many different biological processes including protein function regulation, protein–protein interaction, membrane–protein associations, neuronal development, signal transduction, apoptosis and mitosis (98–100). Dysfunction of palmitoylation has been linked to many diseases including neurological diseases (Huntington’s disease, schizophrenia and Alzheimer’s disease) and different cancers (101–105).
Myristoylation
Myristoylation (N-myristoylation) was discovered by Alastair Aitken in 1982, in bovine brain (34). Although often refers to myristoylation as a PTM, it usually occurs co-translationally (106). This modification is an irreversible PTM that occurs mainly on cytoplasmic eukaryotic proteins. Myristoylation has been reported in some integral membrane proteins as well (107). Myristoylation happens approximately in 0.5–1.5% of eukaryotic proteins (108). In myristoylation after removal of the initiating Met, a 14-carbon saturated fatty acid, called myristic acid, is attached to the N-terminal glycine residue via a covalent bond (Figure 3I) (109). This attachment is often observed in Met-Gly-X-X-X- Ser/Thr motif and is catalyzed by an N-myristoyl transferase (NMT) (there are at least two types of NMT enzymes, NMT1 and NMT2, in humans) (109, 110). Myristoylation occurs more frequently on Gly and less frequently on Lys residues (6).
Proteins that undergo this PTM play critical roles in regulating the cellular structure and many biological processes such as stabilizing the protein structure maturation, signaling, extracellular communication, metabolism and regulation of the catalytic activity of the enzymes (109, 110). The role of myristoylation has been proved in the development and progression of various diseases such as cancer, epilepsy, Alzheimer’s disease, Noonan-like syndrome, and viral and bacterial infections (111).
Prenylation
The first study on prenylation was done in 1978 by Yuji KamiIya et al. in yeast (33). It is another important lipid-based PTM, which occurs after translation as an irreversible covalent linkage mainly in the cytosol (112). This reaction occurs on cysteine and near the carboxyl-terminal end of the substrate protein (113). Prenylation has two main forms: farnesylation and geranylation (114). These two forms contain the addition of two different types of isoprenoids to cysteine residues: farnesyl pyrophosphate (15-carbon) and geranylgeranyl pyrophosphates (20-carbon), respectively. In prenylated proteins, one can find a consensus motif at the C-terminal; the motif is CAAX where C is cysteine, A is an aliphatic amino acid and X is any amino acid (115). This process is catalyzed by three prenyltransferase enzymes: farnesyltransferase (FT) and two geranyl transferases (Figure 3J) (GT1 and GT2) (48).
The prenylation is known as a crucial physiological process for facilitating many cellular processes such as protein–protein interactions, endocytosis regulation, cell growth, differentiation, proliferation and protein trafficking (115–117). Observations showed that disruption in this modification plays crucial roles in the pathogenesis of cancer (114), cardiovascular and cerebrovascular disorders, bone diseases, progeria, metabolic diseases and neurodegenerative diseases (118, 119).
Sulfation
Sulfation was first discovered by Bruno Bettelheim in bovine fibrinopeptide bin in 1954 (120). Residues Tyr, Cys, and Ser have been identified as target residues for prenylated proteins (6). Often, the target residue of this PTM is tyrosine, which happens in the trans-Golgi network. N-sulfation or O-sulfation includes the addition of a negatively charged sulfate group by nitrogen or oxygen to an exposed tyrosine residue on the target protein (121, 122). Currently, PTS is observed mainly in secreted and transmembrane proteins in multicellular eukaryotes and have not yet been observed in nucleic and cytoplasmic proteins (121). This reaction is catalyzed by two transmembrane enzymes, tyrosyl protein sulfotransferases 1 and 2 (TPST1 and TPST2) (30). TPSTs govern the transfer of an activated sulfate from 3-phospho adenosine 5-phosphosulfate to tyrosine residues within acidic motifs of polypeptides (Figure 3K) (121).
Recently, it has been observed that PTS has vital roles in many biological processes like protein–protein interactions, leukocyte rolling on endothelial cells, visual functions and viral entry into cells (123). This PTM involves in many diseases like autoimmune diseases, HIV, lung diseases and multiple sclerosis (12).
Involvement of PTMs in diseases and biological processes
PTMs have a vital role in almost all biological processes and fine-tune numerous molecular functions. Therefore, the footprints of disruption in PTMs can be seen in many diseases. Figure 4A shows a tripartite network of PTM involvement in diseases and biological processes for the 10 abovementioned PTMs. This network contains 97 diseases and 153 biological processes. Panels B and C in Figure 4 show the biological processes with degree ≥3 (those biological processes that interact with at least three different PTMs) and diseases with degree ≥2, respectively.
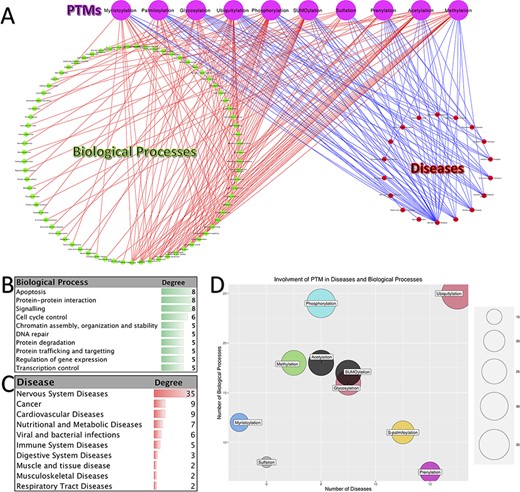
Involvement of PTMs in diseases and biological processes. (A). Tripartite network of PTM involvement in diseases and biological processes for the 10 major PTMs. (B) The degree of the biological processes with degree ≥3 in the tripartite network. (C) The degree of the diseases with degree ≥2 in the tripartite network. (D) Involvement of PTMs in disease and biological processes.
As it is shown in Figure 4C, neurodegenerative disease is the major group of diseases, which is affected by the disruption in the PTMs (Alzheimer’s disease, Parkinson’s disease and Huntington’s disease). Besides, one can see that cancer is also one of the most affected diseases. Consistently with this observation, the biological processes related to cancer are among the high-degree nodes (signaling, DNA repair, control of replication and apoptosis). Processes related to apoptosis, protein–protein interaction, signaling, cell cycle control, chromatin assembly, organization and stability, DNA repair, protein degradation, protein trafficking and targeting, regulation of gene expression and transcription control are the other high-degree biological processes. Moreover, we can say that ubiquitylation, prenylation, glycosylation, S-palmitoylation and SUMOylation have the most involvement in diseases. On the other hand, the PTMs with the highest number of interactions with biological processes are phosphorylation, ubiquitylation, methylation, acetylation and SUMOylation. Putting all together, we can conclude that the disruption in the pathways of these five PTMs has a great impact on the normal functioning of the cell and, as the result, on the organisms
Main PTM databases
Due to the considerable cost and difficulties of experimental methods for identifying PTMs, recently many computational methods have been developed for predicting PTMs (124). Almost all of these methods need a set of experimentally validated PTMs to build a prediction model. Therefore, the availability of valid public databases of PTMs is the first step toward this end. There are a variety of such public databases that could be utilized easily by the scientific community for developing computational methods (17, 124).
According to the scope and diversity of the covered PTMs, these databanks can be classified into two main groups: general databases and specific databases. The general databases contain different types of PTMs, regardless of target residue and organisms. These databases provide a broad scope of information for various PTMs. On the other hand, specific databases have been created based on some certain types of PTMs, certain characteristics of PTMs and/or specific target residues.
The current public PTM databases are greatly different in the number of stored modified proteins, the number of modified sites and the number of covered PTM types. Figure 5 shows a bubble chart of main PTM databases according to these three parameters. As it is evident from the figure, due to the extensive number of studies on phosphorylation, the specific databases are mainly focused on phosphorylation. From this point of view, glycosylation is the second most interested PTM. In the following, the five largest databases are described briefly. Also, Table 1 summarizes the current main public PTM databases.
Major databases for PTMs
| General statistics | ||||||
|---|---|---|---|---|---|---|
| Acronym | Number of covered organisms | Number of PTM types | Number of PTMsa | Type of data and databaseb | URL | |
| General database | dbPTM | More than 1000 organisms | 130 | S: ∼908 900 P: ∼557 700 | Exp. and Pred. Secondary | http://dbptm.mbc.nctu.edu.tw |
| BioGRID | 71 organisms | 6 | S: ∼700 000 P: ∼419 400 | Exp. Primary | https://orcs.thebiogrid.org | |
| Phosphosite Plus | 26 organisms | 7 | S: ∼483 700 P: ∼20 200 | Exp. Primary | https://www.phosphosite.org | |
| PTMCode v2 | 19 organisms | 69 | S: ∼316 500 P: ∼ 45 300 | Exp. Secondary | http://ptmcode.embl.de | |
| qPTM | Human | 10 | S: ∼296 900 P: ∼19 600 | Exp. Secondary | http://qptm.omicsbio.info/ | |
| PLMD | 176 organisms | 20 | S: ∼285 700 P: ∼53 500 | Exp. Secondary | http://plmd.biocuckoo.org | |
| CPLM | 122 organisms | 12 | S: ∼189 900 P: ∼45 700 | Exp. Secondary | http://cplm.biocuckoo.org | |
| YAAM | Saccharomyces cerevisiae | 19 | S: ∼121 900 P: ∼680 | Exp. Secondary | http://yaam.ifc.unam.mx | |
| HPRD | Human | 9 | S: ∼ 93 700 P: ∼30 000 | Exp. Primary | http://www.hprd.org | |
| PHOSIDA | 9 organisms | 3 | S: ∼80 000 P: ∼28 700 | Exp. Secondary | http://www.phosida.com | |
| PTM-SD | 7 model organisms | 21 | S: ∼10 600 P: ∼842 | Exp. Secondary | http://www.dsimb.inserm.fr/dsimb_tools/PTM-SD | |
| WERAM | 8 organisms | 2 | S: ∼ 900 P: ∼584 | Exp. Secondary | http://weram.biocuckoo.org | |
| Phosphorylation | EPSD | 68 organisms | Phosphorylation | S: ∼1 616 800 P: ∼209 300 | Exp. Secondary | http://epsd.biocuckoo.cn |
| PhosphoNET | Human | Phosphorylation | S: ∼950 000 P: ∼20 000 | Exp. and Pred. Secondary | http://www.phosphonet.ca | |
| RegPhos | Human, mouse and rat | Phosphorylation | S: ∼111 700 P: ∼ 18 700 | Exp. and Pred. Secondary | http://140.138.144.141/∼RegPhos | |
| Phospho.ELM | Mainly model organisms | Phosphorylation | S: ∼42 900 P: ∼8600 | Exp. Secondary | http://phospho.elm.eu.org | |
| Phospho3D | Mainly model organisms | Phosphorylation | S: ∼42 500 P: ∼8700 | Exp. Secondary | http://www.phospho3d.org | |
| dbPSP | 200 prokaryotic organisms | Phosphorylation | S: ∼19 300 P: ∼8600 | Exp. Secondary | http://dbpsp.biocuckoo.cn/indExp.php | |
| pTestis | Mouse | Phosphorylation | S: ∼17 800 P: ∼3900 | Exp. and Pred. Secondary | http://ptestis.biocuckoo.org | |
| LymPHOS | Human | Phosphorylation | S: ∼15 500 P: ∼4900 | Exp. and Pred. Primary | https://www.lymphos.org | |
| P3DB | 9 plant organisms | Phosphorylation | S: ∼14 600 P: ∼6400 | Exp. and Pred. | http://www.p3db.org | |
| Glycosylation | UniPep | Human | N-glycosylation | S: ∼52 400 P: ∼40 100 | Exp. and Pred. | http://www.unipep.org |
| GlycoFly | Drosophila melanogaster | N-glycosylation | S: ∼740 P: ∼477 | Exp. and Pred. | http://betenbaugh.jhu.edu/GlycoFly | |
| GlycoFish | Zebrafish | N-glycosylation | S: ∼269 P: ∼169 | Exp. and Pred. Primary | http://betenbaugh.jhu.edu/GlycoFish | |
| Ubiquitylation | mUbiSiDa | 7 model organisms | Ubiquitylation | S: ∼110 900 P: ∼ 35 400 | Exp. Secondary | http://reprod.njmu.edu.cn/mUbiSiDa |
| S-palmitoylation | SwissPalm | 17 organisms | S-palmitoylation | S: ∼1062 P: ∼664 | Exp. and Pred. Secondary | https://swisspalm.org |
| S-nitrosylation | dbSNO | 18 organisms | S-nitrosylation | S: ∼4200 P: ∼2200 | Exp. Secondary | http://dbSNO.mbc.nctu.edu.tw |
Number of PTM sites and modified proteins, which are abbreviated as S and P, respectively
‘Type of data’ can be experimental and/or predicted, which are abbreviated as Exp. and Pred., respectively. ‘Type of database’ can be primary or secondary. A database was considered as secondary if it was an integration of some other databases.
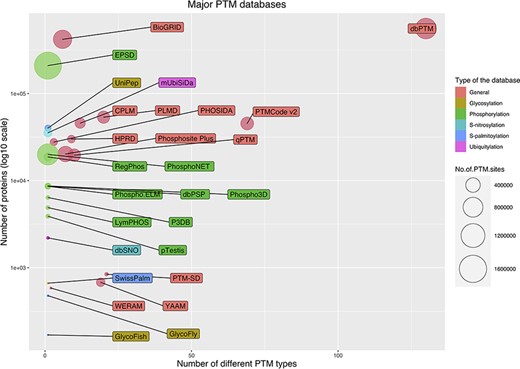
Bubble chart for PTM databases. The chart was drawn based on three parameters for the databases: the number of stored modified proteins, the number of modified sites and the number of covered PTM types.
The EPSD (Eukaryotic Phosphorylation Site Database) contains the largest number of PTM sites. EPSD contains more than 1 600 000 experimental phosphorylation sites in more than 209 000 phosphoproteins across 68 eukaryotes, including 18 animals, 7 protists, 24 plants and 19 fungi (125).
dbPTM (Database Post-translational modification) is a comprehensive database that has collected experimental PTMs’ data from 30 public databases and 92 648 research articles. dbPTM contains ∼908 000 experimentally verified sites for more than 130 types of PTMs from different organisms (6). This database is the largest database in terms of the number of recorded proteins and also in terms of the number of stored PTM types (Figure 5).
BioGRID (The Biological General Repository for Interaction Datasets) is another major open access PTM database. In addition to protein and genetic interactions, it also holds data on ∼726 000 phosphorylation sites in ∼ 72 000 proteins, which were extracted from 4742 publications for 71 major model organisms (126).
PSP (PhosphoSitePlus) is an online resource for studying experimentally observed PTMs such as phosphorylation, ubiquitinylation and acetylation. PSP is comprised of ∼484 000 PTM sites for more than 7 PTM types from 26 species. However, the major amount of its data are extracted from human, mouse and rat (127).
The qPTM database contains 10 types of PTMs for ∼296 900 sites in more than 19 600 proteins under 661 conditions that are collected and integrated into a database (128).
Computational methods for predicting PTMs
Generally speaking, any computational method for predicting a specific type of PTM has four main steps: data gathering, feature extraction, learning the predictor and performance assessment. These steps have been schematically shown in Figure 6. In the following, these steps are described in detail. Also, the related challenges and problems in each step are discussed as well.
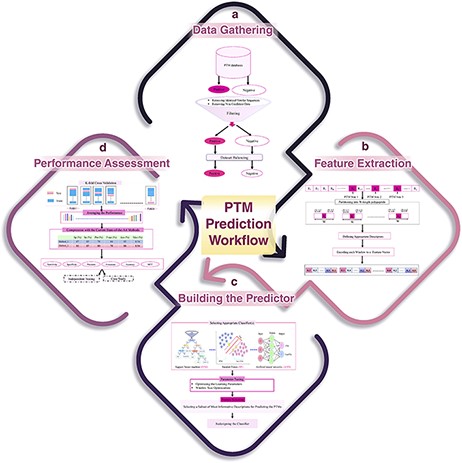
A schematic flowchart to show how a predictor works for PTM prediction. (A) Data collection and dataset creation. (B) Feature selection. (C) Creating training and testing models. (D) Evaluation of the performance of the models.
Data gathering
The first step of a PTM prediction method is gathering the data of proteins that undergo the PTM of interest, in order to assemble a valid dataset (Figure 6A). The final dataset must include both positive (polypeptide sequences having a target residue that has undergone PTM) and negative (polypeptide sequences having a target residue that has not been affected by PTM) samples in order to enable us to train a machine learning algorithm for predicting PTMs.
Positive data selection: almost all studies use the aforementioned databases (such as dbPTM or Uniprot) to gather the positive samples.
Negative data selection: selecting the negative dataset is the most challenging part of the data gathering step. There are three main strategies for selecting the negative dataset.
A random set of proteins with an equal number of the positive set is selected. Then, those occurrences of the target residue that did not undergo the PTM are considered as the negative samples.
The second strategy works like the first, but only those proteins are considered, to construct the negative dataset, that none of their target residues have undergone that specific PTM (based on experimental evidences).
The third strategy examines only the proteins that are included in the positive dataset. In this case, those occurrences of the target residue that have not undergone PTM are considered as the negative samples.
Filtering
After constructing the primary positive and negative datasets, one important task is removing inconsistent/redundant samples to gain a more reliable dataset. This step varies from study to study. One can distinguish three main policies in the literature for removing inconsistent/redundant proteins:
Removing identical proteins
Removing similar proteins within the positive and negative datasets
Removing similar proteins between the positive and negative datasets.
CDhit (129) is used as the major tool to detect similar samples (sequences). However, the threshold of identity for considering a pair of sequences to be similar/redundant varies across different studies. This threshold varies between 40% and 100% in different PTM prediction studies (130).
Dataset balancing
Regardless of the strategy that is used for the negative data selection, in almost all cases, filtered datasets are imbalanced, and size of the negative dataset is greater than that of positive dataset in various extent (sometimes the negative dataset is greater by some order of magnitude). Due to the biases that can be introduced by the imbalanced datasets in the learning phase (when a very specialized learning method is not used, which usually is the case), prior to the feature extraction and learning a classification model, a dataset balancing step is required. To have a balanced positive/negative dataset, often, a random subset of the negative dataset with an equal number of samples to the positive samples is selected.
Feature extraction
In this step, the positive or negative samples (protein sequences), according to the various biological properties, are coded into numerical feature vectors to be used to learn the final predictor (classifier). For this encoding, firstly, using a sliding window, all proteins are partitioned into polypeptides with length W, in such a way that the target residue (according to the PTM of interest) is placed at the center of the polypeptides (Figure 6B). W is an odd number, and therefore, (W − 1)/2 residues are placed on the left and right sides of the target residue in each polypeptide (a window of W residues). There is no agreement on the size of W, and various sizes have been used in different studies. Roughly speaking, W varies from 11 to 27. Some studies select an optimized size for W through a try-and-error approach (130). Finally, according to the appropriate biological descriptors such as amino acid composition, di-peptide composition, similarity score to the known motifs and physicochemical properties, each polypeptide of length W is encoded as a numerical feature vector.
Learning the predictor
After feature extraction, data are ready to train a classifier (model) for predicting the PTM, given a protein of interest (Figure 6C). There are a variety of classifiers that can be trained. At this step, based on the performance of different classifiers and knowledge of the experts that are involved in the study, a suitable classifier is selected. After parameter optimization, the classifier is trained on a subset of the assembled dataset (that is called the training dataset), and then, the predictor is ready to be assessed and compared with the current state-of-the-art methods. In some studies, an additional process, named feature selection, is done prior to building the final predictor. In feature selection, a subset of the most informative/discriminative features are selected and used to learn the classifier.
Performance assessment
k-fold cross validation
A standard and widely used procedure for assessing the performance of a given classifier is k-fold cross validation (Figure 6D). In this process, the available dataset is randomly partitioned into k equal-sized disjoint subsets. Then, k − 1 of the subsets is used as the training dataset, and the remaining one is used as the test set for evaluating the predictor. This process is repeated k times in such a way that every subset is used exactly once as the test set. Finally, the average performance over all k test sets is reported. The most common values for k are 5 and 10 in the PTM prediction studies. Despite the fact that some studies have used a large value for k, the large values lead to less accurate estimates of the generalization power of the classifier and test error rate (131). The most important performance assessment measures that are used in the PTM prediction methods include sensitivity (Recall), specificity, accuracy, precision and Matthews’s correlation coefficient. All of these measures can be calculated based on the four basic elements of the confusion matrix (Table 2). For definition of these performance, refer to Refs. (132, 133).
Confusion matrix for PTM prediction tools
| Experimentally validated PTMs | |||
|---|---|---|---|
| PTM | No PTM | ||
| Prediction | PTM | TP (true positive: number of real PTM sites that have been truly predicted as PTMs) | FP (false positive: number of real non-PTM sites that have been wrongly predicted as PTMs) |
| No PTM | FN (false negative: number of real PTM sites that have been wrongly predicted as non-PTMs by the predictor as PTMs) | TN (true negative: number of real Non-PTM sites that have been truly predicted as Non-PTMs) | |
In addition to the aforementioned measures, ROC and area under the ROC curve are also two major performance evaluation measures (132).
Common flaws in performance assessment via k-fold cross validation procedure
There are some important flaws in performance comparison based on k-fold cross validation, which can lead to a biased conclusion. As mentioned above, the data are randomly portioned into k distinct folds (subsets) in a k-fold CV procedure. Therefore, if only the train and test data of all the k folds are identical for two methods, the results of those methods are comparable. However, many studies compare their k-fold CV results without satisfying this condition. Another common flaw is using the same data for parameter tuning (and feature selection) and for performance evaluation. In such situation, the performance of the predictor is overestimated, and the classifier will perform poorly on the unseen samples.
Independent test
In the presence of enough data for the PTMs, which usually are available except for newly discovered PTMs, some studies carry out an independent test experiment. In this experiment, a dataset of positive and negative samples is assembled (or a benchmark dataset may be used) as an independent test data, which have not been used in any of the previous steps, and the performance of the classifier is evaluated again using this dataset. Usually, the performance on an independent test set is lower than that of k-fold CV and is a better estimation of the real-world performance of a method. To show the strength of the proposed methods in real-world biological problems, some studies use their trained models on a set of biologically important proteins, which have recently been studied, to indicate that their method can effectively detect the newly reported and experimentally validated PTMs.
Tools for PTM prediction
Considering the high cost of experimental identification of PTMs, in recent years, many computational methods have been proposed for the prediction of PTMs. Many of these methods have been introduced as publicly accessible tools. Figure 7 provides a comprehensive list of these tools. In addition to the PTM prediction tools, Nickchi etal. proposed the ‘Post-translational modification Enrichment Integration and Matching Analysis’ (PEIMAN) software for carrying out PTM enrichment analysis on proteins (26). PEIMAN is a publicly accessible standalone software (http://bs.ipm.ir/softwares/PEIMAN/) that uses the UniProtKB database to extract PTM terms. In addition to the enrichment analysis, PEIMAN also performs a comparative analysis. In this case, PEIMAN gives two distinct lists of proteins and then integrates the enrichment results and provides a list of highly enriched terms of both protein sets.
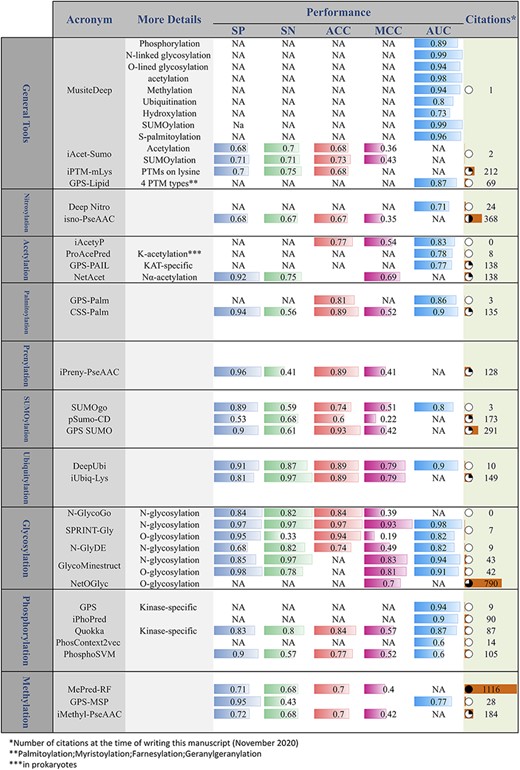
Online PTM prediction tools. The values of five important performance assessment measures have been extracted from the related publications: specificity (SP), sensitivity (SN), accuracy (ACC), Matthews’s correlation coefficient (MCC) and area under the ROC curve (AUC).
Conclusion
PTMs are the chemical modification of a protein after translation and have a wide range of effects on the function and structure of the target proteins. These processes occur on almost all proteins, and many domains within proteins are modified on multiple amino acids by diverse modifications. The function of a modified protein is often strongly affected by these modifications that play important roles in a myriad of cellular processes. There is strong evidence that shows that disruptions in PTMs can lead to various diseases. Hence, increased knowledge about the potential PTMs of a target protein may increase our understanding of the molecular processes in which it takes part. High-throughput experimental methods for the discovery of PTMs are very labor-intensive and time-consuming. Thus, there is an urgent need for prediction methods and powerful tools to predict PTMs. There is a considerable amount of PTM data available from various publicly accessible databanks, which are valuable resources for mining patterns to train new models for PTM prediction. In recent years, many computational methods have been developed for this purpose. However, there are some common weaknesses in assessing these methods, and so it seems that such methods should be evaluated more critically. Considering the diversity of PTMs and new PTMs that are reported every couple of years on one hand, and the advancement of machine learning algorithms on the other hand, we can conclude that this field will attract more attention in the future.
Acknowledgements
The authors would like to thanks Mohammad Hossein Afsharinia for his help with preparing the graphics and Saber Mohammadi for his help with editing the manuscript. Also, the authors appreciate the anonymous reviewers for their very constructive comments.
References
Author notes
Shahin Ramazi and Javad Zahiri contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}