





Abstract
Biocuration plays a crucial role in building databases and complex systems-level platforms required for processing, annotating and analyzing ‘Big Data’ in biology. However, biocuration efforts cannot keep pace with a dramatic increase in the production of omics data; this presents one of the bottlenecks in genomics. In two pathway curation jamborees, Plant Reactome curators tested strategies for introducing researchers to pathway curation tools, harnessing biologists’ expertise in curating plant pathways and developing a network of community biocurators. We summarize the strategy, workflow and outcomes of these exercises, and discuss the role of community biocuration in advancing databases and genomic resources.
Introduction
Big data generation in biology is becoming easier, cheaper and faster. However, scientists need cyber-infrastructure and platforms capable of providing a systems-level framework (genomic context) for visualization and analysis of high-throughput omics data in order to unleash the full potential of the data—which in turn can provide useful insights for translational research (see Figure 1). These systems-level platforms such as Ensembl–Gramene (1), Phytozome (2), Kyoto Encyclopedia of Genes and Genomes (KEGG) (3), Reactome (4), Plant Reactome (5), BioCyc (6) and species-specific metabolic networks [i.e. Plant Metabolic Network (7), RiceCyc (8), MaizeCyc (9), VitisCyc (10), FragariaCyc (11), etc.] allow integration of various automated, semiautomated and/or manual biocuration pipelines, and provide researchers a user-friendly framework based on data mining, back-end data processing and biocuration.
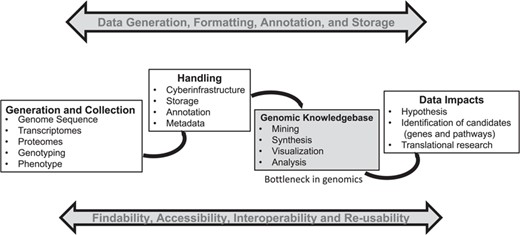
A general summary of the genomics pipeline depicting ‘Big Data’ generation, handling, analysis and its potential translation. The development of cyber-infrastructure and analysis tools is ongoing for storage, analysis and visualization of the raw and processed data simultaneously to make it available for integration across various public platforms using the FAIR (findable, accessible, interoperable and reusable) principle. A genomic knowledgebase such as Plant Reactome represents the downstream end of this pipeline, which can directly support the generation of a data-driven hypothesis for translation of biological knowledge.
Biocuration is a time-intensive process that involves the creation of a user-friendly narrative of biological information based on the review, analysis and systematic organization of biological data from various sources using manual and/or semi-automated methods (12–14). Currently, biocuration of genomic data is one of the factors limiting progress in genomics due to limited financial support for biocuration and a shortage of trained curators (15–18). Powerful gene orthology-based algorithms are available for projecting gene annotations from model species to other species (1, 5, 19, 20), and some progress has been achieved in automated text mining (21–23). However, the reliable alternative of a trained biocurator, who can synthesize a new knowledge by integrating diverse data types, does not yet exist. Engaging and training the researchers and database users in biocuration could be a long-term solution for breaking this barrier and sustaining various biological knowledgebase(s) (18).
Gramene (http://gramene.org) has worked with the community of plant researchers and supported efforts in standardizing and formatting data sets accurately for deposition in the public repositories for long-term utilization, reuse, reanalysis and interoperability (24, 25). In order to engage the community for biocuration of genes and pathways, we have organized two pathway curation jamborees in 2017 and 2018 at Oregon State University. The aim of these jamborees was to introduce plant researchers to biocuration strategies and tools, develop a network of biologists for community curation and harness their expertise for curation and peer review of Plant Reactome pathways.
Here we summarize a successful biocuration jamboree (design, process, workflow and outcomes) and discuss strategies for enhancing the participation of the plant research community in biocuration of genes, proteins and pathways.
Pathway biocuration jamboree: design and implementation
We organized a pathway curation jamboree in connection with the 2018 International Conference on Biological Ontology (ICBO 2018). Results of previous one-on-one work with individual biologists and a small jamboree at 2017 International Gene Ontology Consortium meeting suggested that successful training required a well-defined project, multiple training sessions spread over several days, and a gradual introduction of tasks associated with the curation process to lead participants from familiar chores such as literature review to more specialized aspects of data organization and entry. One week prior to the jamboree, Gramene curators shared a list of 10 pathways (4–5 research articles associated with each pathway) with the participants and offered them the opportunity to add pathways or articles of their choice for curation. A pre-survey to participants was sent to assess their expertise, familiarity with biocuration and expectations from the jamboree. To accommodate the needs of participants who were only interested in the curation process but not in the literature review (or had time commitment conflicts), our curators prepared a list of 300 rice genes associated with abiotic stress pathways.
For the Jamboree itself, we planned a 4-day (20-hour) curation workshop, where participants could participate at their own pace while attending other activities at the conference. We opted for a ‘knowledge-centric’ focus for this workshop (as opposed to ‘tool-centric’). The biocuration exercise began with a simplified approach that employed the strengths of biologists (literature review, gathering data, evaluating evidence and synthesizing knowledge). We then compartmentalized various tasks: enrichment of gene annotations, making gene–gene connections and manual pathway illustration, followed by using PathVisio and the Reactome Curator Tool. We provided individual help in navigating concepts and tools.
Table 1 summarizes the strategy, approach and outcomes of the 2018 Biocuration Jamboree. In the first step, the participants were asked to gather a list of genes and summaries in Excel sheets based on their literature review. We then discussed the importance of standardized gene IDs and instructed participants to resolve gene ID conflicts in the rice genes associated with their assigned pathway.
Strategy, workflow and outcomes of the 2018 pathway jamboree
| 1. Data gathering on excel sheet | |||
|---|---|---|---|
| Data type | Specific task | Curation outcomes | Learning outcomes |
| Literature review (3–4 hr per article) | Research articles on five pathways: Arsenic transport, HSFA7 gene regulatory network involved in drought response, heat response, cold response and endosperm development were selected to identify (i) genes associated with a pathway or biological process and (ii) gene–gene or gene–protein interaction. | From 7 research articles, a list of 200 genes was extracted; 6 gene–gene interactions involved in heat stress were identified; and a network of HSFA7 transcription factor consisting of 35 genes was deduced. | Graduate students found the critical review of literature useful in their own research. |
| Gene IDs conversion (2–3 min per gene) | Participants were instructed to convert old gene IDs (e.g. MSU gene ID) to the standard RAP gene IDs using this tool: https://rapdb.dna.affrc.go.jp/tools/converter. | 196 gene IDs were converted. 4 MSU genes do not have corresponding genes in RAP. | Provided insights into the value of consistency in gene nomenclature and synthesis of knowledge. |
| Transcript IDs (2 min per transcript) | Ensembl transcript IDs were used for depicting transcription events. | 36 transcript IDs were extracted. | Showed how to get transcript Ids. |
| UniProt IDs (5 min per gene) | To acquire a protein IDs and annotation for a specific gene product from UniProt (https://www.uniprot.org). | 189 UniProt IDs were mapped to RAP genes. | Learned that genomic resources are not perfect and are a work in progress. |
| Subcellular location within a plant cell (5–15 min per protein) | The default location of genes and transcripts is nucleus. To assign subcellular location to a protein, experimental evidence from the literature or prediction from the compendium of crop proteins with Annotated locations (cropPAL) (http://crop-pal.org) or TargetP analysis (http://www.cbs.dtu.dk/services/TargetP) was used. | 189 proteins were assigned subcellular location. This provides enrichment of annotation. | Students found these tools to be useful for their own research project. |
| Transmembrane domain(s) (5 min per protein) | TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) was used for prediction of transmembrane helices in proteins. | 26 were found to be transmembrane proteins. | This is a useful exercise to enrich protein annotation. |
| GO (2–3 min per gene) | Assign GO terms | GO terms were assigned to 36 transcription events and in the case of proteins, were extracted from UniProt. | Learned about the utility of ontologies in curation. |
| Molecular interactions (15 min) | Summarized interaction data | Discussed a transcription network of 36 genes and another network of 6 genes, which emerged from literature review. | Learned how false-positive protein–protein interactions can be identified by considering their subcellular location. |
| Reaction (10 min) | Showed how to assign a reaction to a gene and protein. | Metabolic/transport/translocation/transcription/binding events were illustrated on whiteboard. | How different types of reactions can be depicted in pathways |
| Pathway (10 min) | Showed how to assemble various reactions into a pathway and to associate them with complex biological processes (i.e. development of plant organs and tissues, plant’s response to abiotic stress). | As an example, the gene regulatory network of HSFA7 was associated with ABA-dependent drought response. | How to integrate information from various sources to create a pathway. |
| Summary (15–30 min per summary) | Create a summary of genes and pathways with citation | In total, summaries of 20 genes and 5 pathways were composed. | Critical review of data from the literature |
| 2. Visualization of a biological pathway and reactions | |||
| Idea | Activity | Curation outcome | Learning outcomes |
| Discussed how various macromolecular interactions can be depicted in the form of a series of connected reactions (2 hours) | 15 min whiteboard presentation by each participant. | The rough outline of five pathway diagrams was created. | This exercise was highly appreciated by all participants. Beyond biocuration exercise, it helped them in building data-driven hypotheses and refining their own research projects. |
| 3. Drawing pathways and reactions using pathway curation tools | |||
| Tool | Activity | Curation outcome | Learning outcomes |
| PathVisio tool (1 hr) | Showed curation of gene–gene network involved in rice response to biotic stress | The curated pathway was deposited in WikiPathways | Participants found this to be a useful resource. |
| Introduction to Plant Reactome analysis tools and the Reactome Curator Tool (6 hr) | Short introduction to Plant Reactome Introduction to the Reactome Curator Tool by showing the step-by-step curation of an arsenic transport pathway. Participants curated pathways of interest with one-on-one assistance provided by organizers | Curation of arsenic transport pathway. Curation of one gene-regulatory pathway and partial curation of three additional pathways. | Participants found Plant Reactome to be a useful resource for the analysis of large-scale gene expression datasets, and the Reactome Curator Tool to be the most sophisticated pathway curation platform. |
Historically, after the completion of the Oryza sativa ssp. japonica cv. Nipponbare genome sequencing (26), two projects, the Michigan State University Rice Genome Annotation Project (MSU project; http://rice.plantbiology.msu.edu) (27) and the Rice Annotation Project (RAP; https://rapdb.dna.affrc.go.jp) (28) simultaneously led rice gene annotation efforts. At present, the RAP system prevails, but both gene nomenclature systems (gene locus IDs) are in use. At Plant Reactome/Gramene, we use standard RAP gene nomenclature. Thus, to summarize rice literature, a curator often has to first convert an MSU gene ID (e.g. LOC_Os03g44380) into a corresponding RAP gene ID (e.g. Os03g0645900) using a gene converter tool (available at https://rapdb.dna.affrc.go.jp/tools/converter). In the literature, we also find other types of gene IDs in use (i.e. GenBank accession numbers, gene array/probe identifiers, and cDNA), which makes it difficult to map on a standard gene ID, and requires sequence search analysis (e.g. using BLAST).
Furthermore, we showed how to acquire information on the gene product (protein) from UniProt (https://www.uniprot.org). Currently, one rice gene ID corresponds to one or more protein IDs in UniProt. Using the options provided by UniProt’s search feature, we select the best match based on quality criteria (i.e. `gold star’ sets, six-digit IDs, longest proteins and preferably from rice proteome data set). The participants were also instructed on how to acquire additional information on the subcellular location and membrane association of proteins. Gene ontology (GO) annotations for proteins were automatically imported from UniProt into the Reactome Curator Tool. GO annotations to transcripts of 36 genes associated with the heat shock transcription factor HSFA7 gene-regulatory network (29) were added manually.
In the second step, we asked participants to depict various macromolecular interactions in the form of a series of connected or isolated reactions to describe gene–gene interactions. These reactions were then grouped in a pathway and associated with a biological process (illustrated either on paper or using PowerPoint). Each participant was asked to summarize their work and other participants were encouraged to add or suggest modifications. Finally, trained Gramene curators provided their suggestions on how to connect information from one publication to another and extend the gene–gene interaction network or pathway. Where the discreet knowledge of a reaction was not available, event types were depicted as BlackBoxEvents.
Once the participants had gathered the required data and visualized the macromolecular interactions graphically, we began pathway curation using PathVisio (https://www.pathvisio.org), a free, simple open-source application developed by WikiPathways. We encourage researchers to take advantage of training material available at http://academy.wikipathways.org to generate pathway figures and share their knowledge with the community using the WikiPathways platform.
In the last step, we introduced participants to the Plant Reactome database (5) and its analytical capabilities applicable to their research. Finally, we used the Reactome Curator Tool (https://reactome.org/download-data/reactome-curator-tool) to demonstrate step-by-step curation of arsenic uptake and detoxification in rice. The learning curve for the Reactome Curator Tool is typically quite steep and it takes our curators 4–6 months to get acquainted with all aspects of it. By introducing the Reactome Curator Tool in this manner, we were able to focus on the biological content of pathway curation, while still laying the groundwork for future in-depth training on the Plant Reactome platform.
Curation and learning outcomes
In total, 20 participants registered for the pathway biocuration jamboree in 2018, but due to various parallel workshops and other activities in the conference only 8 participated including 1 professor, 1 postdoc, 5 graduate students and 1 research assistant, who devoted 4–20 hours to curating rice genes and pathways. The participants mined genes from 7 published papers, provided detailed annotations for 200 genes and curated one complete regulatory pathway (HSFA7 gene network) and three partial pathways for the Plant Reactome database. The curated data was attributed to their name in the pathway curation tool and the same will be preserved when the data are made publically available. In addition, two professors working only in non-plant species spent time learning pathway curation strategies, but did not contribute to the curation.
Interestingly, one graduate student opted to work on a script that can automatically populate data in an Excel sheet by pulling gene IDs and UniProt IDs. The script (available at https://github.com/mttmartin/uniprot_lookup) does a good job of automatically mapping MSU gene IDs to RAP gene IDs and pulling the corresponding UniProt IDs where only one gene ID is mapped to one UniProt ID. However, it fails to select the appropriate UniProt ID when more than one options are available. This program is useful for automating some aspects of manual pathway curation and can be a part of our ongoing effort to map one gene to one UniProt reference accession for the canonical peptide form. The student’s computational contribution was a useful surprise. We learned that community contribution can come in many forms and workshops such as this can be a collaborative learning space.
Beyond curation for Plant Reactome, the participants found this exercise very useful and directly applicable to their own research. Graduate students, in particular, liked the critical review of literature, identifying well-curated from non-curated data in public databases and appreciated the standardized gene nomenclature concept for an organism. Participants working with non-model organisms also appreciated the concepts of GO, consistency in gene nomenclature and the ability to draw pathways for their organisms in PathVisio to share with the community. The participants also found Plant Reactome and WikiPathways as useful resources for analysis of their own research data (see supplementary data). Scientists frequently use curated information available in various databases. This jamboree provided participants an opportunity to learn about the biocuration process and protocols that are required for building and improving genomic resources.
Community biocuration: challenges and opportunities
As a long-term strategy, community curation can help to build secondary resources at a fast pace. However, significant resources are needed to train researchers in curation before this activity becomes useful and productive. Therefore, existing databases should have a mandate to train the community and allocate resources for such activities. Conference sites are ideal for conducting biocuration workshops and recruiting curators from a large, diverse pool of researchers and also for economizing resources. We were able to conduct two curation workshops at our host institute, in conjunction with the 2017 Gene Ontology Consortium meeting, and again at the ICBO 2018 Conference. Participants in these conferences are acquainted with, or at least interested in, the challenges of handling big data and are actively engaged in the creation of sustainable curation models, policies and tool development. In addition, the parallel workshops on GO, ontology development, data standards, machine learning and natural language processing (NLP) also complemented our curation jamboree. We also had limited resources and time. Therefore, it made sense to conduct a local experiment, where we could provide maximum help to participants. In the future, depending upon the availability of financial resources, we can attempt hosting a similar workshop at a larger conference venue to recruit plant researchers.
The 2017 jamboree participants were mostly senior faculty. We faced the challenge of retaining these participants for a full day (as other workshops/lecture sessions were running in parallel). These participants learned how Plant Reactome could be useful for their research projects but could not commit their full attention to biocuration due to other conference commitments. Learning from this experience, we designed a more flexible schedule for the 2018 jamboree, spread over 4 days. Prior to the jamboree, our curators interacted with local laboratories, learned about their research projects and needs, introduced the utility of Plant Reactome and recruited participants. In addition, we sent a pre-survey form to all registered participants and adapted our strategy and workflow on the basis of their responses. We also sent a video tutorial, a simple pathway curation protocol and a list of selected research articles to registered participants.
In the 2018 Pathway Biocuration Jamboree, we had 50% local and 50% outside registrants. In general, local graduate students committed 50–100% of their time to biocuration, whereas the randomly registered participants did not commit enough time or chose to attend other workshops. The participant graduate students found our approach to a critical review of the literature, hypothesis building, data organization and analysis during biocuration exercises directly applicable to their own research projects. We were surprised by their sincere, dedicated efforts and time investment. The training of the local group of graduate students was accomplished at minimal cost and served as a primer for how best to structure and organize biocuration jamborees for a wider community of researchers.
We suggest that engaging graduate students early on in biocuration activities will lay a foundation for building a community of researchers who can proactively format their research data for easy integration into public databases. If incorporated in the graduate school curriculum, biocuration training could be beneficial to students and simultaneously increase the community’s contribution to biocuration of public databases.
The support and motivation to train and engage biology researchers in biocuration could also come from publishers, and/or from collaborative efforts of public databases, and societies. For example, Scientific Data, a research journal published by Nature Publishing Group, has taken an initiative to engage authors in metadata curation prior to publication and to provide specific templates for furnishing the required information (30).
Conclusion
We benefited tremendously from the participation of active researchers in two pathway curation jamborees. We learned where gaps exist between bioinformatics resources and the needs of researchers. Depending upon the availability of resources and funding, we will continue our efforts to engage the plant genomic community in biocuration and collaborate with other institutions/publicly funded projects in such activities. We look forward to engaging graduate students, postdocs and researchers from local, national and international institutions. We welcome researchers who would like to directly contribute to plant pathway curation. Researchers can curate/edit pathways using PathVisio in WikiPathways and provide a summary with citations. Our curators can import and adapt pathways from WikiPathways to Plant Reactome with lesser effort. Alternatively, researchers can also send pathway drawings and summaries to Plant Reactome curators directly or volunteer as a reviewer for curated pathways. We also welcome one-on-one training (online, at conferences or site visits) depending on the availability of funds.
Gramene in collaboration with CyVerse and the Texas Advanced Computing Center is also working with American Society of Plant Biology (ASPB) to engage authors in data curation. The ASPB’s two high-impact journals The Plant Cell and Plant Physiology have developed an NLP-based biocuration and author engagement workflow that is activated at the time of author proof validation step. This workflow uses Domain Informational Vocabulary Extraction and requests edits and validation from authors (31). This effort is funded by the US National Science Foundation. Such support and structured requirements from publishers will train researchers in aspects of biocuration and would allow for better integration of data from research articles into public databases.
The investment of various stakeholders (academia, industry, educators, scientific societies and publishers) in engaging and training the broader research community in biocuration will provide a sustainable and quality solution for keeping pace with the big data explosion. We hope that Plant Reactome’s efforts in engaging the user community in biocuration can help other databases to effectively strategize and plan similar workshops. Acquiring big data literacy and biocuration skills is an important incentive for researchers. Such exercises could help young researchers to think more critically about their own project, train in using relevant tools and resources and engage with the broader scientific community.
Finally, we agree with the suggestions made earlier by Howe et al. (18) and reiterate that community curation will be sustainable when individuals are appropriately rewarded for their work; granting agencies recognize community curation as an outreach component and institutions include it as an acceptable service component when making tenure decisions. Community curation efforts without the supervision of trained curators are less likely to be productive and sustainable. Professional curators will remain as the backbone of quality biocuration by (i) recruiting, training and supporting the community; (ii) reviewing the work submitted by community curators; (iii) developing new protocols for curation, software development and testing and (iv) outreach.
Acknowledgements
We thank Peter D’Eustachio (Reactome project) for training Plant Reactome curators, providing suggestions on this manuscript and his guidance in organizing the 2017 pathway curation jamboree. We acknowledge technical support provided by Guanming Wu and Justin Elser. We thank all 2017 and 2018 jamboree participants. We thank volunteer community curator, Bijayalaxmi Mohanty, for compiling the list of 75 rice genes that are transcriptionally regulated in response to cold that we used in the curation workshop for writing/testing a script for automatic mapping of MSU gene IDs to RAP gene IDs and corresponding UniProt IDs.
Funding
National Science Foundation (NSF IOS-1127112 to Gramene database project) and National Institutes of Health (P41 HG003751 to Human Reactome project).
Conflict of interest. None declared.
Plant Reactome Database URL: http://plantreactome.gramene.org

{kind=link}