





Abstract
Eukaryotic chromatin is composed of DNA and protein components—core histones—that act to compactly pack the DNA into nucleosomes, the fundamental building blocks of chromatin. These nucleosomes are connected to adjacent nucleosomes by linker histones. Nucleosomes are highly dynamic and, through various core histone post-translational modifications and incorporation of diverse histone variants, can serve as epigenetic marks to control processes such as gene expression and recombination. The Histone Sequence Database is a curated collection of sequences and structures of histones and non-histone proteins containing histone folds, assembled from major public databases. Here, we report a substantial increase in the number of sequences and taxonomic coverage for histone and histone fold-containing proteins available in the database. Additionally, the database now contains an expanded dataset that includes archaeal histone sequences. The database also provides comprehensive multiple sequence alignments for each of the four core histones (H2A, H2B, H3 and H4), the linker histones (H1/H5) and the archaeal histones. The database also includes current information on solved histone fold-containing structures. The Histone Sequence Database is an inclusive resource for the analysis of chromatin structure and function focused on histones and histone fold-containing proteins.
Database URL: The Histone Sequence Database is freely available and can be accessed at http://research.nhgri.nih.gov/histones/.
Introduction
Histones play central roles in both chromatin organization and gene regulation, as they constitute the fundamental protein units of the nucleosome (1). The nucleosome consists of DNA wrapped around an octameric core histone complex, composed of a central H3–H4 tetramer and two adjacent H2A–H2B dimers; the nucleosome is commonly identified as the first order of compaction of eukaryotic chromatin (2). Core histone genes also display conserved expression patterns that show periodic expression across the eukaryotic cell cycle, with a pronounced peak during S-phase (3). This allows for histone proteins to be produced at the same time DNA is being synthesized. Thus, the histone proteins can be readily assembled into nucleosomes and then compacted into chromatin.
Core histones are highly conserved across eukaryotes in terms of sequence and structure. Despite overall sequence conservation, extensive histone tail post-translational modifications, in addition to histone variants present during development, contribute to epigenetic mechanisms that signal transcriptional activation, repression and recombination events. Histone proteins and their variants have an essential function in gene regulation (4–6). Nucleosomes are disassembled at transcriptionally active promoters via histone post-translational modifications (7), specific histone variants are known to mark active promoters and regulatory regions (8), and other variants are involved in the transition between transcriptionally active or silent chromatin (9). In recent years, much progress has been made toward genome-wide profiling of chromatin modifications (10), where histones play critical roles in defining the overall structure and function of chromatin and, by extension, in gene regulation.
The histone fold is a common structural motif shared by each of the four core histones, which mediates interactions between the individual core histones. The histone fold is structurally composed of three α-helices connected by two loops, and this overall architecture allows for heterodimeric interactions between core histones (11). Interestingly, even though each individual histone protein family is highly conserved, the histone fold is not conserved at the sequence level but, rather, at the structural level (12). Higher-resolution crystal structure of the nucleosome core particle has demonstrated detailed structures of the histone folds in each of the histones (13, 14). The DNA wrapped around each nucleosome is held in place by linker histones (called H1, or H5 in avian species). The linker histones, which do not contain the histone fold motif and have a different evolutionary origin from the core histones (15), are critical to chromatin higher-order compaction and facilitate internucleosomal interactions (16). In addition, H1 variants have been shown to be involved in the regulation of developmental genes (17). The overall structural state of chromatin controls DNA replication, recombination and gene expression, with histones playing critical roles during these processes (18).
Interestingly, despite the conservation of core histone gene expression patterns, the regulatory machinery that controls core histone gene expression has changed greatly among eukaryotic evolutionary lineages. Specifically, the identity of the core histone gene cis-regulatory sequence motifs and the protein factors that bind these motifs are distinct for the yeast Saccharomyces cerevisiae, as well as for other fungi, plants, insects and mammals (19). Therefore, different species have developed unique gene regulatory mechanisms for core histone genes that converge in the same gene expression phenotype, high expression levels specifically during S phase, concomitant with DNA replication.
Although the core histones are among the most slowly evolving eukaryotic proteins, members of the histone H2A and H3 families have diversified extensively, assuming specialized roles in DNA repair, gene silencing, gene expression and centromere function (5, 6). Interestingly, the centromere H3 variant appears to form tetrameric nucleosomes that induce positive supercoils, and these specialized ‘centromeric nucleosomes’ have been proposed as the epigenetic inheritance mechanism for centromeres (20).
The histone fold motif—common to all core histones—has also been found in a variety of non-histone proteins. The large majority of these non-histone proteins are localized in the nucleus and their functions are related to DNA metabolism; they include nuclear factor Y (NF-Y) and the TFIIB transcription factors (12). A few histone fold-containing proteins localized in the cytoplasm include the Ras activator Son of Sevenless (SOS) (21): SOS1 is localized primarily in the nucleus and SOS2 localized in the cytoplasm (22). Huntingtin interacting protein M (CXorf27) also contains a histone fold and is localized in the cytoplasm. We hypothesize that histone folds in cytoplasm-localized proteins are used to mediate protein–protein interactions.
Given the central role of histones and related proteins in a wide variety of critical cellular functions, we feel the need to continue to provide a centralized, curated source of important information on these proteins to the biomedical community. To this end, the Histone Sequence Database represents an organized collection of all histones and histone fold-containing proteins (23). The information presented in this Database includes a list of published three-dimensional structures for histones and histone fold-containing proteins, as well as manually curated multiple sequence alignments for each histone family.
Database and software
Data tables
The Histone Sequence Database, which has been developed and expanded significantly since its last release (23), has three tables stored in a relational database schema using Oracle 10 g (Figure 1). The HISTONES table stores information about the histone category, its accession, the sequence string, the submitting database, as well as NCBI's taxonomic information on the sequence. The ORGANISM table contains detailed taxonomic information for the sequences contained in the Histone Sequence Database. The STRUCTURES table stores information on the experimentally determined structures of proteins contained in the database, including the method of determination (i.e. X-ray crystallography or NMR spectroscopy).
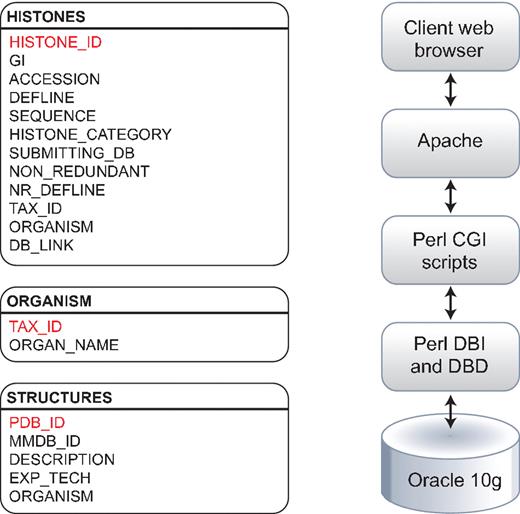
Histone Database data model. The Histone Database stores selected manually curated information from GenBank records. The information stored as part of each record includes the GenBank unique identifier (GI), accession number, definition line, sequence string, histone class, database source, NCBI taxonomic identifier and organism name. The database front-end is written in Perl, the data is stored in an Oracle 10 g relational database, and data is retrieved using Perl DBI and DBD libraries.
Software
The Histone Sequence Database uses Common Gateway Interfaces (CGIs) written in the Perl programming language that communicate with the relational database software. The connectivity between the CGIs and the Oracle 10 g Relational Database Management Software was implemented using Perl's Database Interface (DBI) and the Oracle database driver for the DBI module (DBD::Oracle), available through the Comprehensive Perl Archive Network (CPAN; http://search.cpan.org/). The use of object-oriented design methodologies and Perl modules that are both open source and developed in-house allows for flexibility and scalability. The Web pages displaying data, such as the summary of contents, non-redundant sets, and search pages are dynamically generated using CGI. Comments concerning the Web front-end are welcomed and encouraged.
Data sources and histone protein identification
The protein databases searched for the update and curation of the Histone Sequence Database were the NCBI non-redundant (nr) database (18 November 2010); nr includes sequences of all non-redundant GenBank CDS translations (24), as well as the sequences of RefSeq proteins, sequences of structures represented in the Protein Data Bank (PDB) (25), and sequences from UniProtKB/Swiss-Prot (26), the Protein Information Resource (PIR) (27), and the Protein Research Foundation (PRF) (http://www.prf.or.jp/index-e.html). The collection of histones was extended and revised, using the HMMER3 software package (28). We constructed hidden Markov models (HMMs) for each of the four core histones and the linker histone H1 from the alignments generated in the last release of the Histone Database. Additional HMMs were generated for archaeal histones (29) and bacterial proteins that contain a histone-likefold (30); only the protein entries that have a complete domain hit with an E < 0.01 are collected for further analysis. For each histone family, multiple sequence alignments were generated using MUSCLE (31). The alignments that are manually curated to include proteins with complete folds are also available in PDF format and are color-coded to allow easy identification of amino acid variants. The Histone Database uses a color scheme designed to highlight the specific amino acid differences that a particular group of sequences may have inside the core or linker histone alignments by coloring amino acids with similar physicochemical properties differently. A summary table of the number of sequences found grouped by family and species represented in the database is provided (Table 1).
Histone Database content
| Core histone profile | Number of unique sequences | Increase since last update (%) (23) | Number of unique taxonomic identifiers |
|---|---|---|---|
| H1/H5 | 591 | 138.3 | 156 |
| H2A | 1016 | 214.6 | 331 |
| H2B | 755 | 161.2 | 308 |
| H3 | 2287 | 476.1 | 7096 |
| H4 | 341 | 181.8 | 344 |
| Archaeal | 182 | – | 89 |
Identification of histone fold-containing proteins
Histone fold-containing proteins were identified using a different search strategy. We used the sequences from each of the four core histone MUSCLE alignments (H2A, H2B, H3 and H4) as seeds for PSI-BLAST (32) searches. The PSI-BLAST searches were run to convergence with an E-value inclusion threshold of 0.01; the core histone seeds were excluded from the final list of histone fold-containing proteins. Additionally, related structures were identified using NCBI's VAST-related structures searches (33, 34), in an effort to identify more distant histone fold-containing proteins that could not be identified through PSI-BLAST searches. Using this strategy, we were able to identify a total of 2180 histone fold-containing proteins.
Results and Discussion
The computational approach presented here has identified proteins throughout a wider evolutionary spread of genomes. Currently, the Histone Database contains entries that represent a total of 7356 unique NCBI taxonomic identifiers, which correspond to approximately the same number of organisms. The sequences of core histones, linker histones and archeal histones are available in FASTA format. They are also available as a series of multiple sequence alignments, one for each class of proteins. A number of search engines can be used to query the database in several different ways: by protein family, organism, keyword or based on a sequence pattern (Figure 2). Each histone sequence for which three-dimensional structure data is available is linked to the corresponding entry in both PDB and the Molecular Modeling Database (MMDB) (35).
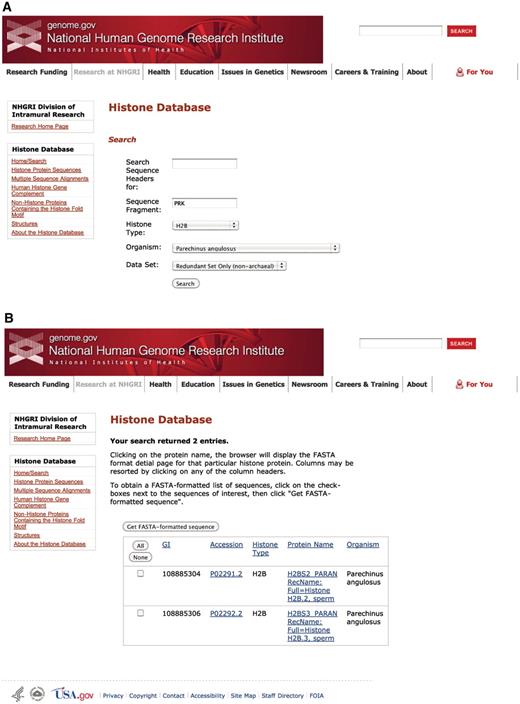
Histone Database query and results. The Histone Database main page displays a search engine that allows users to find histone sequences from a large variety of organisms. Additionally, users have the possibility of exploring other features to access complete collections of Histone Protein Sequences, Multiple Sequence Alignments, The Human Histone Gene Complement, Non-Histone Proteins Containing the Histone fold Motif and Histone Structures. The upper panel shown (A) presents the criteria used for the query, which requires the sequence to contain a fragment with amino acids PRK from the angulate sea urchin (Parechinus angulosus) histone H2B. The search results presented in the lower panel (B) include two protein sequences that meet the criteria specified by the query.
The Histone Sequence Database has been expanded significantly since its last update (23) (Table 1). However, the expansion is not proportional for each of the core histones. The H3 sequences, which contain a large number of variants with specialized roles in chromosome segregation and transcription, show an increase over 400% since the last database update. Similarly, the H2A core histone sequences that include variants with specialized functions in DNA repair and transcription regulation show an increase over 200% since the last update. In contrast, we observe a more modest growth in sequence numbers for the relatively invariant H4 and H2B core histones.
The Histone Sequence Database now includes archaeal histone sequences. The current update contains 182 sequences from 89 archaeal organisms, which includes members of all classified archaeal phyla (i.e. euryarchaeota, crenarchaeota, nanoarchaeota, korarchaeota and the newly proposed phylum thaumarchaeota). The presence of histone folds in all classified archaeal phyla indicates that the histone fold originated before the archaeal and eukaryotic lineage divergence (29). Most of the archaeal histones have a single histone fold domain; however, there are a number of sequences that contain two histone folds, with the C-terminal histone fold sharing higher sequence similarity with archaeal histones with a single histone fold. Archaeal histones containing two histone folds have been proposed as intermediates between archaeal and eukaryotic histones (36, 37), where both core histones H3 and H4 would have originated at the same time, followed by a second event that gave rise to core histones H2A and H2B. In the current release of the Histone Sequence Database, archaeal histones with two histone folds are confined to two distinct branches: Halobacteriaceae and the hyperthermophilic methanogen Methanopyrus kandleri. Although archaeal histones containing two histone folds have been previously identified in these lineages, it is not clear how these histones could also contribute to pack DNA in extreme high temperature or high salinity environments.
Structural comparisons confirmed the presence of the histone fold in the extreme bacterial thermophile Aquifex aeolicus (30). Additionally, the RIKEN Structural Genomics/Proteomics Initiative (RSGI) (38) has solved two Thermus thermophilus structures for a protein that also contain the histone fold (PDB:1WWI and PDB:1WWS). The histone fold was also found in diverse types of bacteria, including aquificales, ε-proteobacteria, thermaceae, actinobacteria and nostocaceae. This suggests that the histone fold appeared in bacteria by lateral gene transfer (29, 39). Interestingly, the structure from T. thermophilus (PDB:1WWS), predicted to be a dimer, is strikingly similar to the H3–H4 tetramer. However, an analysis of the electrostatic surface potential for protein Aq_328 from the hyperthermophilic bacterium A. aeolicus (PDB:1R4V) and archaeal histone from M. kandleri (PDB:1F1E) (Figure 3) reveals the DNA binding surface in the archaeal histone (Figure 3F) but shows no conservation of any of the DNA-binding residues present in both archaeal and eukaryotic histones (Figures 3A and 3D) (29). Therefore, it is possible that histone fold-like bacterial proteins have functions unrelated to DNA binding. However, it is likely that the histone-like fold is used as a dimerization domain in these species.
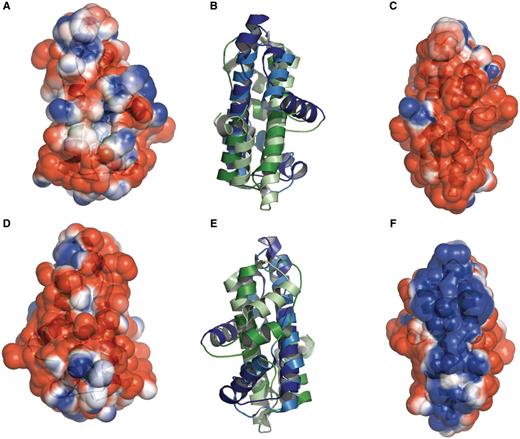
Histone-like folds in A. aeolicus and M. kandleri. Protein Aq_328 from the hyperthermophilic bacterium A. aeolicus (PDB:1R4V) and archaeal histone from M. kandleri (PDB:1F1E) have two histone like folds. These are colored as dark blue and dark green (for 1R4V) and light blue and light green (for 1F1E). The electrostatic surface potential ranges from +2 kTe−1 (blue) to −2 kTe−1(red). (A) and (D) the front and back views, respectively, of the electrostatic surface potential of Protein Aq_328. (B) and (E) superimposed structures of protein Aq_328 and the archaeal histone from M. kandleri. (C) and (F) the front and back views, respectively, of the electrostatic surface potential of the archaeal histone from M. kandleri. The figures were generated with PyMOL (49) and the APBS plug-in for PyMOL (50).
Conclusions
Researchers studying chromatin structure and function have traditionally relied on the Histone Sequence Database to explore the taxonomic breadth of histones and their variants (40–43). Others have focused on epigenetics and transcriptional regulation and use the database to discover newly reported core histones and histone-fold-containing proteins (44–48). The Histone Database continues to be a comprehensive bioinformatic resource that organizes and stores histone sequences and groups them into families (that now includes archaeal histones), maintains a collection of histone fold-containing sequences, and provides information on three-dimensional structures available in PDB. In the future, we will enhance our histone fold identification pipeline with state-of-the-art sequence- and structure-based methods to continue to identify new members of this biologically critical family of proteins. We also plan to integrate functional information from other publicly available Web resources.
Funding
Funding for open access charge: The Intramural Research Programs of the National Center for Biotechnology Information, National Library of Medicine and the National Human Genome Research Institute, both at the National Institutes of Health.
Conflict of interest. None declared.
Acknowledgments
The Histone Database update utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Maryland. (http://biowulf.nih.gov/).

{kind=link}
{kind=link}
{kind=link}