





Abstract
The Mouse Genome Database, the Gene Expression Database and the Mouse Tumor Biology database are integrated components of the Mouse Genome Informatics (MGI) resource (http://www.informatics.jax.org). The MGI system presents both a consensus view and an experimental view of the knowledge concerning the genetics and genomics of the laboratory mouse. From genotype to phenotype, this information resource integrates information about genes, sequences, maps, expression analyses, alleles, strains and mutant phenotypes. Comparative mammalian data are also presented particularly in regards to the use of the mouse as a model for the investigation of molecular and genetic components of human diseases. These data are collected from literature curation as well as downloads of large datasets (SwissProt, LocusLink, etc.). MGI is one of the founding members of the Gene Ontology (GO) and uses the GO for functional annotation of genes. Here, we discuss the workflow associated with manual GO annotation at MGI, from literature collection to display of the annotations. Peer-reviewed literature is collected mostly from a set of journals available electronically. Selected articles are entered into a master bibliography and indexed to one of eight areas of interest such as ‘GO’ or ‘homology’ or ‘phenotype’. Each article is then either indexed to a gene already contained in the database or funneled through a separate nomenclature database to add genes. The master bibliography and associated indexing provide information for various curator-reports such as ‘papers selected for GO that refer to genes with NO GO annotation’. Once indexed, curators who have expertise in appropriate disciplines enter pertinent information. MGI makes use of several controlled vocabularies that ensure uniform data encoding, enable robust analysis and support the construction of complex queries. These vocabularies range from pick-lists to structured vocabularies such as the GO. All data associations are supported with statements of evidence as well as access to source publications.
Introduction
Mouse Genome Informatics (MGI) is the primary international database resource for the laboratory mouse, providing integrated genetic, genomic and biological data to facilitate the study of human health and disease. The MGI team curates the biomedical literature (11 000 publications per year) and normalizes and integrates sequence and functional data about mouse genetics and genomics from almost 50 other external database and informatics resources. MGI organizes curation teams around particular types of data including sequence data, phenotypes, embryonic expression data, comparative and functional information, mouse tumorigenesis and mouse models for human diseases. MGI utilizes multiple bio-ontologies and is the authority for mouse gene and strain nomenclature.
Five projects contribute to this resource. The ‘Mouse Genome Database’ (1) includes data on gene characterization, nomenclature, mapping, gene homologies among mammals, sequence links, phenotypes, disease models, allelic variants and mutants and strain data. The ‘Gene Expression Database’ (2) integrates different types of gene expression information from the mouse and provides a searchable index of published experiments on endogenous gene expression during development. The ‘Mouse Tumor Biology (3) Database’ provides data on the frequency, incidence, genetics and pathology of neoplastic disorders, emphasizing data on tumors that develop characteristically in different genetically defined strains of mice. The MGI group is a founding member of the ‘Gene Ontology Consortium’ (GO, www.geneontology.org, (4)). MGI fully incorporates the GO in the database and provides a GO browser for access to mouse functional annotation. Finally, the ‘MouseCyc’ database (5) focuses on ‘Mus musculus’ metabolism and includes cell level processes such as biosynthesis, degradation, energy production and detoxification. It is part of the BioCyc collection of pathway databases created at SRI International (6).
Here, we outline the workflow process for ‘one’ component of the MGI data acquisition and integration process—that associated with the ‘Gene Ontology Project at MGI (Figure 1)’. MGI assigns functional annotations (GO terms) to genes and protein products through semi-automated methods and manual curation. Semi-automated annotation strategies include mapping and translating data from the Enzyme Commission, Swiss-Prot, InterPro (see http://www.geneontology.org/GO.indices.shtml), rat and human ortholog experimental data and others. Curation of these data sets includes review and resolution of Quality Control Reports generated through the process of data loading and comparisons to existing data in MGI. For the purpose of this paper, we will not discuss these semi-automated integration methods, but rather concentrate on the manual literature curation, which is a vital source of experimental mouse functional data. While we focus on the GO component of MGI in this description, the literature curation process is very similar for other MGI components that curate literature.
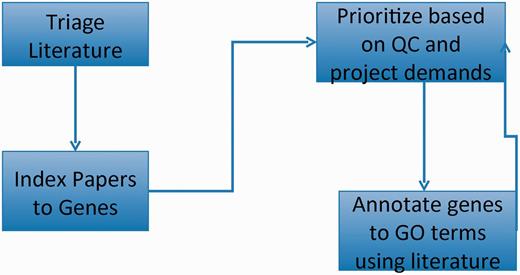
GO curation workflow: papers of interest are identified and entered into the database system (triage) and associated with genes (indexed). GO annotations are made using papers selected based on quality control reports and projects. The quality control reports in turn are revised daily based on added annotation.
The subcomponents of the literature curation process include the following:
- (a)
Literature triage: identifying and obtaining relevant scientific literature
Each curator is assigned a specific subset of journals from a set of 160 relevant journals out of ∼650 subscriptions carried by the Jackson Laboratory. Journals are chosen for triage based on the numbers of articles that have been identified and curated for that journal over the previous years. The manuscripts accepted for curation come from a variety of sources. Most are selected from the journals that are regularly triaged. Others are selected on the basis of particular annotation processes such as full curation of the Wnt family of proteins, or in another example, full curation of genes implicated in human diseases. On a yearly basis, the number of papers selected from all journals is tabulated, and the selection of which journals to regularly curate the next year is determined based on the relevancy of the journal publications during the previous year and the number of full time equivalents available for this task. Typically, a few journals are dropped and a few are added each year to the formal triage process. The QUOSA application (http://www.quosa.com) is used as an aid to access and identify recent papers as represented in PUBMED containing data about the mouse and determining which component of the database will be curated from the paper (GO, expression, mutant alleles, phenotypes, mapping, tumor). QUOSA is used to retrieve full text PDFs for a journal issue or time frame of interest. Because the experimental organism ‘mouse’ is often not mentioned in the abstract (3–34% depending on the journal), a curator selects an issue of a journal and then searches the full text of papers containing the keywords ‘mouse’, ‘murine’ or ‘mice’. The application highlights these terms showing the context of the search keywords, which enables curators to quickly determine whether the experiments described are of a suitable nature to be used for GO annotation (i.e. do the experiments aid in determining the normal function of the gene?) (Figure 2). The number of papers examined and the number selected are somewhat journal dependent. For example, out of ∼80–90 papers per each weekly issue of Journal of Biological Chemistry, roughly 60 contain one or more of the three keywords, and of those, the curator may select 10–15 as being relevant for some area of the database. For an issue of Nature, which may have 15–20 research articles per issue, up to 5 may have the keywords and all of them are relevant. Papers selected are uploaded to an in-house server for the next steps.
- (b)
Adding the publications to the MGI system through the editorial interface
Papers selected as containing data appropriate for GO annotation are entered into a ‘master bibliography’ module. Figure 3a shows the module’s data entry screen. Each record is tagged for the area of use of the database for which it has information. Information about journal, volume, pages and the abstract are automatically obtained nightly from PUBMED using the PMID. The tag for the database area is added manually. At this stage, the paper is not associated with any specific gene. The area tag will automatically change when the paper is actually used for curation. For example, if this paper was used for a GO annotation, the ‘used’ box would then have an X added automatically.
- (c)
Indexing the papers to determine the genes being studied
A paper is then associated or indexed to the genes discussed within by adding the paper to each gene’s detail module (Figure 3b). PROMINER, a natural language processing (NLP) application, is used to assist in the gene indexing process (7). Utilizing official nomenclatures and synonym lists for mouse/rat/human gene names and gene symbols, PROMINER marks up papers for review by a curator, who then associates genes-to-papers in the MGI editorial interface (EI) (Figure 4).
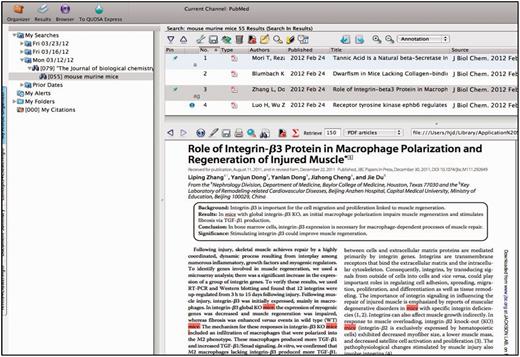
QUOSA Information Manager showing part of a paper from an issue of JBC with mouse, murine or mice highlighted. Curator quickly looks at context to select appropriate area of MGI that the paper best fits. The paper shown has been ‘tagged’ for alleles and GO.
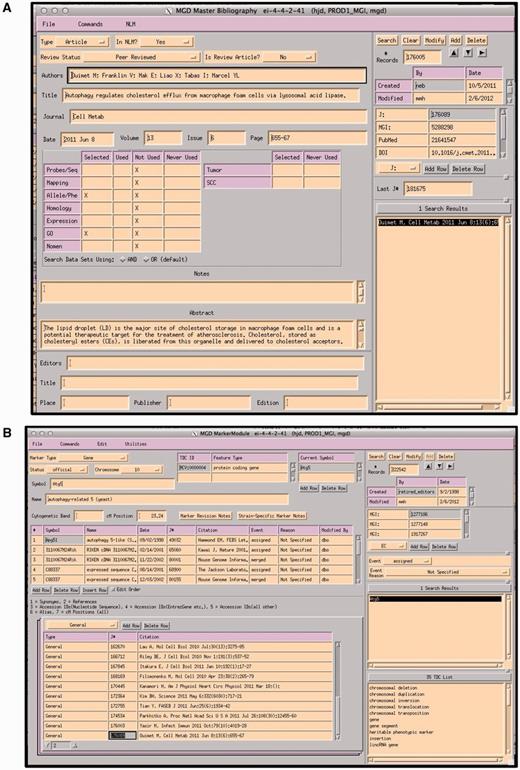
(A) MGI Master Bib EI, showing a record for a paper that has been selected for alleles/phenotypes and GO. The paper has not yet been curated for GO (indicated by an X in the selected and X in Not Used). (B) MGI Gene Feature Detail Module. References to be associated with this gene are entered into the lower left hand panel. If a paper is associated with multiple genes, the same paper is entered for each gene. All EIs are TeleUSE graphical user interface applications running under X-windows.

Output from PROMINER: PROMINER is used to assist in the gene indexing process. Utilizing official nomenclatures and synonym lists for mouse/rat/human gene names and gene symbols, PROMINER marks up papers for review by a curator, who then associates genes-to-papers in the MGI EI.
Sometimes a paper discusses several genes, but not all of them may be objects for direct GO annotation. For example, a paper describing the effects of a knock out of a particular gene may use analysis of other gene products to analyze the particular processes being affected, but the annotation to involvement in the process would only be made to the gene being knocked out. Currently, the topical areas selected for each paper are not directly tied to the genes associated with the paper. Thus, a paper selected for GO for one of the genes will appear in the GO EI interface of unused papers for each of the genes indexed to the paper.
Curation triage: selecting what genes to annotate
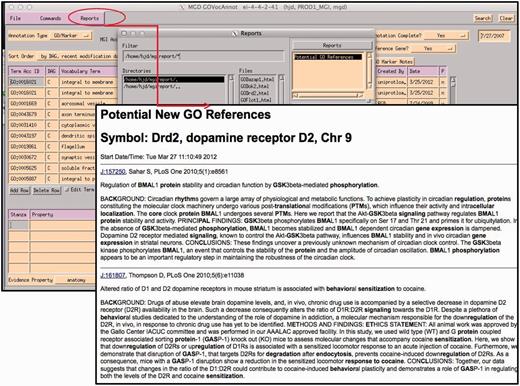
Ideally, all papers would be immediately used for GO annotation, but on average 300 new papers are added to the database each week, only less than half of these are curated each week due to resource limitations. Therefore, various priority selection criteria are used to choose which genes and papers warrant immediate attention. Reports of interest are generated, such as ‘genes with no GO annotation but have new indexed literature selected for GO’ or ‘genes with mutant alleles that have literature selected for GO’ (Figure 5). Additionally, participation in various collaborative projects, such as the Reference Genome project (8), or the Protein Ontology (8) defines primary sets of genes to work based on community input.

GO QC reports used for annotation triage and quality control.
Data entry: creating a GO annotation using the EI module for GO
A curator at MGI uses the GO EI module to enter annotations (Figure 6). Curators use the annotation guidelines set forth by the Gene Ontology Consortium (http://www.geneontology.org/GO.annotation.shtml). The MGI interface is gene centric. It is divided into two main data entry sections: the annotation area and the annotation properties area. A list of papers selected for GO that are associated with this gene is shown in the lower right panel. Individual protein isoforms or modified forms can be indicated using annotation properties where an id for a specific isoform can be indicated. After reading a paper, the curator selects the appropriate GO id (1). Next, the reference number is added (2), as well as the evidence code (3). If required by the type of evidence code, additional information is added to the inferred from column (4). Once the annotation is saved, information about the cell type that the experiment was done in, or the specific isoform, or tissue, is entered into the annotation properties section (5). Additional ontologies such as the Cell Ontology (9), Mouse Adult (10) and Embryonic Anatomies (11), Protein Ontology (12), and psi-Mod (13) are used in the properties fields. Several of these are used to supply an extension to the annotation which are used in the gene association file (GAF) (5). Curators can use the OBO-EDIT tool (14) to load multiple ontologies to aid in searching for appropriate terms, as well as viewing the chosen term in the context of the rest of the ontology (Figure 7). The data entry module has several built-in features to aid in QC. The GO vocabulary is refreshed daily from the GO site, and only current GO terms can be used, otherwise data entry is prohibited. Only reference identifiers previously entered into ‘master bibliography’ can be used. Incorrect evidence codes are automatically rejected. There are other mechanisms, such as data loads, that provide GO annotation. In all cases, provenance is provided. The GO EI also has a built-in report generator that highlights words matching GO terms found in the abstracts of papers selected for GO as an aid to suggesting the type of information and evidence present in a paper (Figure 8).
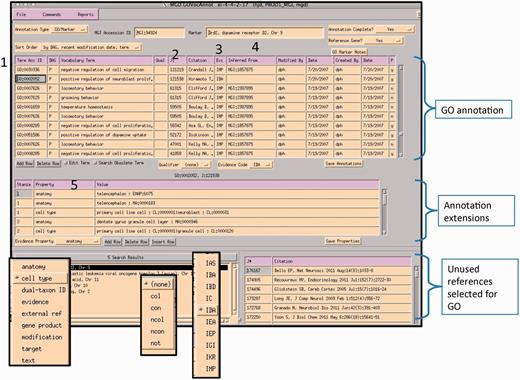
MGI GO data entry module: the interface is divided into three main sections: GO annotation, annotation properties and search and reference tracking. Drop-down menus display pick-lists of allowed entries in various fields (evidence property, GO qualifier and evidence codes). Numbered areas: 1, GO ID entry; 2, reference entry; 3, evidence code entry; 4, ‘inferred_from’ entry required for certain evidence codes and 5, annotation properties entry.
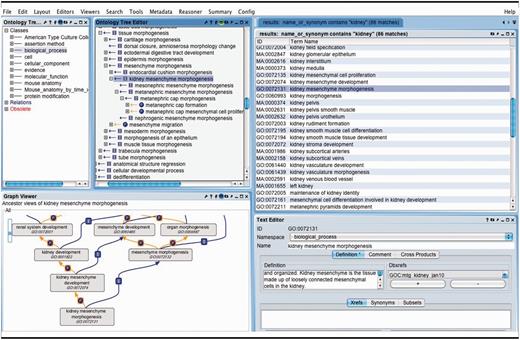
OBO-Edit ontology tool used to browse multiple OBO ontologies. The far left panel shows the vocabularies that have been loaded for searching and viewing. The right panel displays the terms in all of the vocabularies that contain the word ‘kidney’. The GO term ‘kidney mesenchyme morphogenesis’ is selected and is visible as a tree view showing its children (middle panel), and as a graphical view showing its parents (lower left).
Report generated using the abstracts of papers selected for GO for the gene being annotated within the GO EI. Text contained in GO terms in each abstract is highlighted.
Tracking metrics and quality control measures to set priorities for upcoming work
GO annotation metrics in MGI are generated daily. MGI GO curators add on average 200 new annotations per week. Annotations are tracked based on a variety of criteria such as annotation source (MGI curation or data load) and evidence (experimental or predictive, such as through orthology or functional domain). Scripts review changes to the GO structure and provide QC reports for curators noting genes whose annotations may be affected by these changes (Figure 9). Additionally, we use the master bibliography tables and the GO annotations to keep track of various areas that need focus, such as ‘genes with no GO annotation but have papers that are selected for GO but not used’.
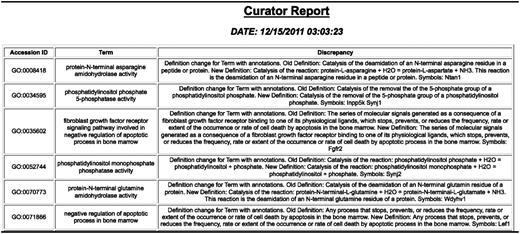
GO change log report showing changes to the GO and genes with annotations using the term that may need to be looked at.
Annotation presentation and usage
GO data for each gene at MGI are displayed to the public in a ‘GO Summary’ page. This page displays the GO annotations as a table, summary text or graph. Sample views for the gene Drd2 are shown in Figure 10. All data assertions in MGI are supported by evidence and citation to the source of the information. For assertions that are associated with controlled vocabularies such as the GO, links are provided to vocabulary browsers that provide the relationships between the assertion and other knowledge in that area of the ontology. Using the table and associated information, MGI provides an automatically generated text description of the GO annotations. MGI also provides a graphical display of GO annotations from the GO detail page for each gene.
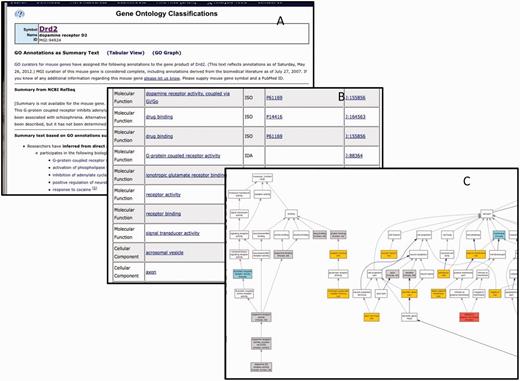
The GO annotation details for Drd2 displayed as summary text (A), table (B) or graph (C). Only a portion of each summary is shown. There are 175 annotations total.
GO annotations are also shared with the GO Consortium (GOC) through a GAF. This is a tab-delimited file that contains most of the elements of a GO annotation as outlined in the GO EI section above. Presently, only the ‘cell type’ and ‘gene product’ annotation properties are included in the GAF. More will be included over time. This file is available on either the GOC web site or along with other data sets, from the MGI FTP site (ftp://ftp.informatics.jax.org/pub/reports/index.html). The GAF and the GO vocabulary file are used as input for many analytical tools such as GO TermFinder (15). Instructions for construction of a GO GAF file are found in GO documentation at http://www.geneontology.org/GO.format.annotation.shtml.
Information access: NLP and beyond
In general, GO annotation from the mouse experimental literature can be very challenging. Although some groups have used NLP to expedite the curation of literature (16), this can be especially difficult to do when applied to mouse biology because of the integration of human and mouse studies within the same description of results. The concepts captured by the GO cannot be gleaned just from simple text matching of terms, but must also take into account inferences that reflect a given context. Additionally, an understanding of the nature of an experimental assay is important to correctly use the information as evidence of a particular result. While we continue to work with NLP developers to design a system to automate identification and tagging of papers (17), it is clear that the complexity of understanding the information in a biomedical publication requires the intervention of an experienced biologist-curator in the process.
Funding
Funding for open access charge: MGI database resources are funded by grants from the National Human Genome Research Institute (HG00330, HG02273), National Institutes of Health/National Institute of Child Health and Human Development (HD062499) and the National Cancer Institute (CA89713).
Conflict of interest. None declared.
Acknowledgements
The Mouse Genome Informatics group are Mark Airey, Anna Anagnostopoulos, Randal P. Babiuk, Richard M. Baldarelli, Jonathan S. Beal, Dale A. Begley, Susan M. Bello, Judith A. Blake, Carol J. Bult, Donna L. Burkart, Nancy E. Butler, Jeffrey Campbell, Lori E. Corbani, Howard Dene, Alexander Diehl, Mary E. Dolan, Harold J. Drabkin, Janan T. Eppig, Jacqueline H. Finger, Kim L. Forthofer, Peter Frost, Sharon Giannatto, Jill R. Lewis, Terry F. Hayamizu, David P. Hill, James A. Kadin, Debra M. Krupke, Michelle Knowlton, Monica McAndrews, Susan McClatchy, Ingeborg McCright, David B. Miers, Howie Motenko, Steve Neuhauser, Li Ni, Hiroaki Onda, Janice Ormsby, Jill Recla, Deborah J. Reed, Beverly Richards-Smith, Joel E. Richardson, Martin Ringwald, David Shaw, Robert Sinclair, Dmitry Sitnikov, Constance M. Smith, Cynthia L. Smith, Kevin Stone, John Sundberg, Hamsa Tadepally, Monika Tomczuk, Linda Washburn, Jingjia Xu and Yunxia Zhu.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}