





Abstract
While alternative splicing (AS) can potentially expand the functional repertoire of vertebrate genomes, relatively few AS transcripts have been experimentally characterized. We describe our detailed manual annotation of vertebrate genomes, which is generating a publicly available geneset rich in AS. In order to achieve this we have adopted a highly sensitive approach to annotating gene models supported by correctly mapped, canonically spliced transcriptional evidence combined with a highly cautious approach to adding unsupported extensions to models and making decisions on their functional potential. We use information about the predicted functional potential and structural properties of every AS transcript annotated at a protein-coding or non-coding locus to place them into one of eleven subclasses. We describe the incorporation of new sequencing and proteomics technologies into our annotation pipelines, which are used to identify and validate AS. Combining all data sources has led to the production of a rich geneset containing an average of 6.3 AS transcripts for every human multi-exon protein-coding gene. The datasets produced have proved very useful in providing context to studies investigating the functional potential of genes and the effect of variation may have on gene structure and function.
Database URL:http://www.ensembl.org/index.html, http://vega.sanger.ac.uk/index.html
Introduction
The alternative splicing (AS) of RNA transcripts can produce multiple mature transcripts from a single locus and the vast majority of human multi-exon loci are subject to AS (1, 2). AS is observed at a high frequency in vertebrates (3), but has also been shown to occur in invertebrate, plant and fungal genomes (4–7). It is believed that AS can act to expand the protein repertoire of the cell (8) encoding alternative protein forms with biological functions that differ from the canonical product of the locus (9). However, AS transcripts may contain profound changes to the structural and functional domains of the canonical product (10), and the loss or disruption of such domains may render the translation product non-functional (11). Instead, it has been proposed that certain non-translated AS transcripts may play a role in gene regulation (12–14). For example, the generation of AS transcripts containing premature termination codons has been shown to regulate the translational output of members of the SR gene family (15), whereby such transcripts become targets for the nonsense-mediated decay (NMD) pathway (16).
Capturing the detail and complexity of AS is important in providing context to any analysis that relies on knowledge of the position, structure and functional potential of gene loci. Our manual annotation of the ENCODE pilot regions (17) was demonstrably more accurate than any computational method in terms of the sensitivity and specificity of models produced (18, 19) and these qualities remain high in current annotation (Harrow, J. et al., submitted for publication, Howald, C. et al., submitted for publication). In collaboration with the GENCODE consortium, we are producing the reference gene annotation for the ENCODE project (20) and this publicly available geneset has also been used by the 1000 Genomes Loss of Function project (21) among others.
Overview of manual annotation of AS
We annotate all transcript models supported by experimental evidence, predominantly ESTs and mRNAs. Manual annotation is well suited to interpreting issues of sequence quality that can compromise the ability of many computational gene builders to utilize ESTs, extracting useful information from them using alignment visualisation tools such as Blixem and Dotter (22, Barson,G. and Griffiths,E., manuscript in preparation). As such, there is no quality-based pre-filtering of ESTs and mRNAs on the basis of e.g. library or tissue of origin, rather every single piece of transcript evidence is considered on its own merits. Where conventional transcriptional data such as ESTs and mRNAs are unavailable, we utilize other evidence to support the annotation of a transcript model, for example, experimental data from publications, conservation in other species, and data from new technologies such as RNA-Seq data and proteomic data. The evidence used to construct each transcript model is made viewable to the user via the Vega (23) and Ensembl (24) browsers. Furthermore, our manual approach allows us to construct transcript models with higher information content than is possible via automated annotation. First, we provide a carefully considered interpretation of the functional potential of the model; e.g. whether the transcript has a full-length CDS, is subject to NMD or non-stop decay (NSD) (25), or has a retained intron that would disrupt the CDS. Secondly, we tag any non-standard features possessed by the model with standard ontologies; for example, where the model contains non-canonical splice sites supported by cross-species conservation.
We avoid extrapolation wherever possible. To avoid both combinatorial inflation of AS transcripts and the assignment of potentially incompatible AS events in the same transcript, we do not merge shorter pieces of evidence (such as ESTs) to create a full-length transcript model unless there is absolutely no other evidence (mRNA or protein) that spans the full-length of the locus. Similarly, to avoid over-predicting the functional potential of an AS transcript we do not flag models as NMD or NSD where the true end of the transcript cannot be identified due to 3′-truncation. Nonetheless, we annotate models even in the absence of interpretable function. For example, we have annotated models that lack a CDS or polyadenylation features from the earliest days of the human genome project (26). However, it is only recently that such transcripts have been recognized as belonging to a class of long non-coding RNA (lncRNA), the putative regulatory function of which are only starting to be confirmed in the laboratory (27). With ∼10 000 annotated lncRNA gene loci containing ∼16 000 transcripts, the GENCODE geneset currently contains the largest set of manually annotated lncRNAs available (Derrien,T. et al., submitted for publication). It is for similar reasons that we annotate the retained intron class of AS variants despite questions over their functional relevance. While retained intron events have traditionally been interpreted as immature or incorrectly processed RNA species captured during cytoplasmic RNA preparations, there is evidence from both bioinformatic (28) and laboratory studies that a subset of such transcripts may be functional. However, due to the continued uncertainty over their validity [e.g. whether they disproportionately derived from tumour cell lines (29)], and in order not to add contamination to protein databases, we do not annotate AS transcripts with retained introns as protein-coding unless there is additional evidence to do so.
Methodology for the identifying and biotyping alternative splice variants
Manual annotation of AS is performed according to the guidelines of the HAVANA (Human And Vertebrate Analysis and Annotation) group; the current set can be accessed at ftp://ftp.sanger.ac.uk/pub/annotation. Tran-script models are built based on the alignment of transcriptomic (ESTs and mRNAs) and proteomic data from GenBank and Uniprot. These data are aligned to the reference genome sequence using BLAST (30), with a subsequent realignment of transcript data by Est2Genome (31). Gene models are manually interpreted from the alignments by annotators using the otterlace annotation interface (32). Alignments are navigated using the Blixem alignment viewer (22). Visual inspection of the dot-plot output from the Dotter tool (22) is used to resolve any alignment with the genomic sequence that is unclear or absent from Blixem. Short alignments (<15 bases) that cannot be visualized using Dotter are detected using the Zmap DNA Search pattern-matching tool (33). The annotation of exon-intron boundaries requires the presence of canonical splice sites (after (34) but defined as GT–AG, GC–AG and AT–AC donor and acceptor sites) and any deviation from this rule requires the use of clear explanatory tags. It is important to note that models are only extended to the extent of the homology with supporting evidence; for example an AS model based on a 3′-truncated EST will not be extended to cover the full length of the locus. Any models based on truncated evidence are clearly tagged to indicate this.
All non-redundant, multi-exon alignments of transcriptional evidence at an individual locus are used to build transcript models. Single exon, unspliced ESTs are not used to construct new transcript models, but may be used to extend the final exon of a model where they support the annotation of polyadenylation features; similarly, unspliced mRNAs can be used to extend the final exon of a model or to build novel, single exon transcript models. All AS events described in Figure 1 are annotated; exon skipping (single or multiple exons), intron retention, alternative splice donor site (5′- and 3′-shifts), alternative splice acceptor [5′- and 3′-shifts, including NAGNAG (35)], alternative first exon, alternative final exon and mutually exclusive exon pairs. Although it is an important concept in describing AS, we do not routinely define a reference transcript at a locus or classify the nature of the AS event. AS is not limited to one event per transcript and transcripts may contain multiple AS events.
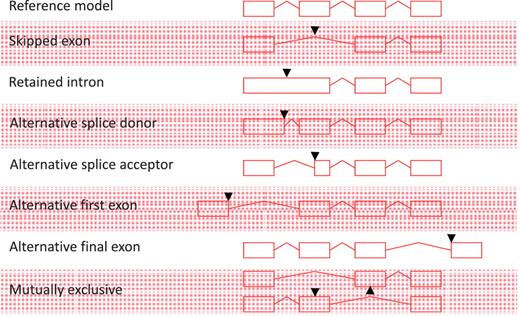
Alternative splicing events. All possible individual alternative splicing events are shown. Black arrowheads indicate position of difference with a conceptual reference model (top).
As part of the GENCODE consortium we work closely with computational collaborators to produce the reference human geneset for the ENCODE project. To ensure the highest possible sensitivity and specificity are maintained, manual annotation is both informed by, and checked against, computational predictions of alternatively spliced transcripts by PASA (36) and Ensembl (37, 38), supported introns (Mark Diekhans, personal communication), U12 introns from U12DB (39), coding exons by CONGO (40) and pseudogenes by PseudoPipe (41, 42), Retrofinder (43) and Pseudofinder (44). Computational gene predictions are visible in the annotation interface to provide hints to annotators during first-pass manual annotation and also compared to completed manual annotation to identify potential missing features and flag them for manual re-investigation. Annotated gene models are validated by the high-throughput sequencing of pooled RT–PCR reactions from eight tissues (brain, heart, kidney, liver, lung, spleen, skeletal muscle and testis) where primers are designed to check single or multiple exon-exon junctions (designated as RT–PCR-Seq) (Howald,C. et al., submitted for publication).
Once their exon-intron structure is resolved, all AS transcripts are assigned to a subclass based on their putative functional potential and structural properties. These subclasses are designated ‘biotypes’ as they aim to reflect biologically relevant features of the transcript. The protein-coding potential of the transcript is initially determined on the basis of similarity to known protein sequences, or homology to orthologous and paralogous proteins. Further information to aid classification may be drawn from the presence of Pfam functional domains (45) possible alternative ORFs, retained intronic sequence and polyadenylation features. Significantly, we also classify the transcripts as putatively susceptible to NMD and NSD. In summary, we explicitly link the structural impact of an AS event to its effect on the functional potential of a transcript, enriching the annotation at both the transcript and locus level (46). For example it is useful to know whether a transcript with a single skipped exon retains an intact CDS or is subject to a frameshift leading to the incorporation of a premature stop codon likely to induce NMD. AS variants at lncRNA loci are predominantly classified on the basis of known non-coding function and positional relationship to protein-coding loci (see Supplementary Figure 1 for more information on assignment of biotypes at protein-coding and lncRNA loci).
Incorporating automatically derived transcripts into the manual geneset
To maintain high sensitivity and specificity, manual annotation is by necessity a slow process compared to computational gene prediction; a full first pass of the human genome has not yet been completed. As such GENCODE utilize Ensembl automated gene predictions as placeholders until a locus or AS transcript can be manually annotated. Currently ∼85% of the transcripts in the GENCODE geneset are manually annotated, with the proportion increasing with every release. Current technologies utilising short read data such as RNA-Seq (1) and RT–PCR-Seq (Howald,C. et al., submitted for publication) are likely to be useful in revealing and validating new AS events. While there is great interest in adding AS transcripts based on these data, the short read lengths generally only allow one or two introns to be captured by a single read. Paired end reads can go some way to help, but in longer genes a lack of connectivity between reads makes it very difficult to deconvolute all possible combinations of AS events to identify those which actually occur together. Building complete transcript models from such data is likely to remain problematic for the foreseeable future (http://www.GENCODEgenes.org/rgasp/). As such, we currently only accept split reads (i.e. reads mapping to two exons) that validate an individual intron, using them to support an AS transcript. We do not use gene predictions constructed by assembling a cluster of reads into a full-length transcript as support for building manually annotated gene models. Nonetheless we anticipate that longer sequencing reads generated by new technologies such as those being developed by Pacific Biosciences (http://www.pacificbiosciences.com) and Oxford Nanopore (http://nanoporetech.com) will eventually provide connectivity between AS events to provide a complete set of full-length AS transcripts for both protein-coding and non-coding loci. In addition, the advent of targeted proteomics approaches (47) combined with higher resolution mass spectrometry has some potential to enrich the set of AS transcripts and validate protein-coding potential.
Complexity of publicly available genesets
As a consequence of the approach we take to annotation i.e. incorporation of ESTs and cross-species evidence, the GENCODE geneset contains a large amount of AS compared to other genesets. The GPCR56 locus encoding human G protein-coupled receptor 56, for example, has 76 AS variants (Figure 2). The GENCODE v7 geneset contains 20 687 protein-coding loci with 122 909 transcripts. Excluding single exon loci, protein-coding loci have a mean of 6.31 transcripts, of these 2.59 are annotated with coding potential and 1.8 give rise to a novel CDS. The difference between the AS transcripts with coding potential and those encoding a novel CDS indicates the large numbers of AS observed in 5′-UTRs (and to a much lower extent 3′-UTR). Approximately 60% of AS transcripts are not annotated with a CDS due to our conservative approach to prediction of protein-coding potential; for example where the transcript retains intronic sequenceor has novel first or last exons within coding introns, and we lack certainty that the true transcription start site or termination site has been captured.
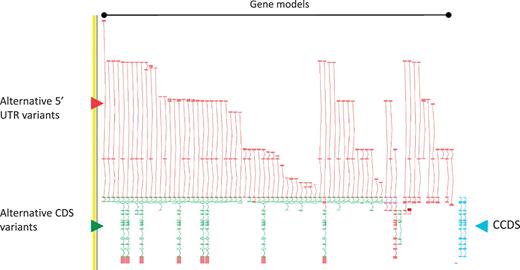
Zmap screenshot of the GPCR56 locus encoding human G protein-coupled receptor 56. This locus has 76 non-redundant AS transcripts, the majority of which are EST based, 3′ truncated and show variation in the 5′ UTR. Red arrowhead indicates 5′ UTR, green arrowhead indicates CDS and blue arrowhead indicate the two CCDS variants annotated at this locus.
A comparison of the GENCODE geneset with the RefSeq geneset (which, like GENCODE, contains genome-wide annotation of protein-coding loci with a significant manual annotation component) reveals the number of protein-coding loci in each set to be similar (20 687 versus 23 191). The difference that does exist is likely due to the fact that the GENCODE geneset is genome-centric whereas the RefSeq geneset is transcript-centric; the latter can also include loci that cannot be mapped to the current reference genome (GRCh37). The total number of loci in the Consensus coding sequence (CCDS) geneset (48), which contains CDSs agreed by Sanger, RefSeq and UCSC, is 18 167, comparable to the individual GENCODE and RefSeq protein-coding genesets. However, While the total numbers of protein coding loci are similar for these sets, the numbers of AS transcripts annotated vary greatly between the genesets with GENCODE containing ∼3.3-fold more AS transcripts than RefSeq and ∼5.2-fold more than CCDS (and is still ∼1.4-fold higher when only AS transcripts with novel CDS sequences are considered).
While protein-coding loci are enriched for AS it is important to note that, in our annotation, AS is not restricted to this biotype. In the GENCODE geneset lncRNA loci commonly have annotated AS transcripts, with a mean of 1.6 transcripts per lncRNA locus. Notable examples include H19 (with 14 variants), the human ortholog of mouse Rosa26 (20 variants) and GAS5 (29 variants), the latter being particularly enriched for retained-intron variants that could potentially play a role in regulating the snoRNA that lie within its introns (Figure 3).
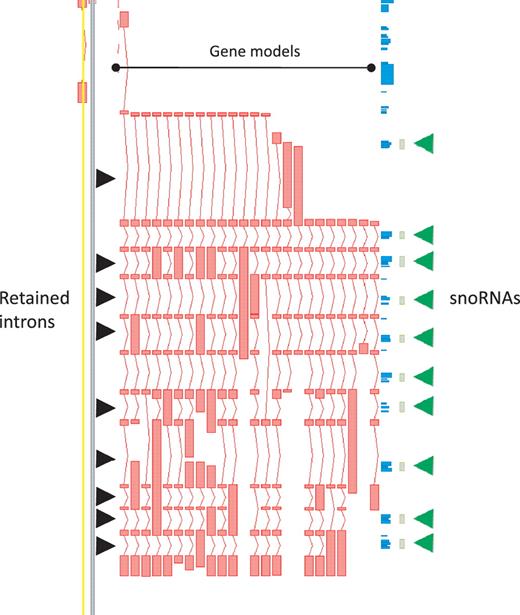
Zmap screenshot of the GAS5 locus encoding growth arrest-specific 5 (non-protein coding). This locus has 29 non-redundant AS transcripts, including many containing retained introns. Black arrowheads indicate those introns retained in at least one transcript. Green arrowheads indicate intronic snoRNAs.
Validation of AS using conservation and transcription as proxies
While the existence of widespread AS is not disputed, the overall contribution of AS to phenotype remains a topic of great debate (11). To date, the vast majority of AS transcripts lack published evidence for their functionality, and even fewer have had their cellular roles defined in the laboratory. This is, at least in part, because the common biochemical assays used to explore functionality are low-throughput techniques, typically better suited to single gene studies than to genome-wide surveys. Even demonstrating the existence of AS protein products in vivo is problematic due to the low coverage achieved by high-throughput techniques such as Mass Spectrometry (49). As such, we must rely on proxies to assess genome-wide functionality for the immediate future. For example, comparative studies may show that a particular AS event is observed in different species; where conservation is observed over significant evolutionary time it can then be hypothesized that the AS event is functional. Recent studies in our group have found that ∼15% of AS events are conserved between human and mouse (45). For example, the PI4KB/Pi4kb genes which encode phosphatidylinositol 4-kinase, catalytic, beta in human and mouse respectively share an exon skipping event that leads to the utilisation of an internal translation start (Figure 4). In addition, we have begun to incorporate data from new sequencing technologies (e.g. RNA-Seq) to judge the functionality of AS transcripts via two main routes. First, if an AS transcript is consistently identified to be present at a significantly lower level than a known functional transcript it is considered likely to be the product of a noisy splicing event; if it is found at a similar level it appears more likely to be functional. Secondly, differential tissue expression patterns can identify putative functional transcripts; an AS transcript that is consistently present in one or more tissue types but absent from others, even when present at a low level, is likely to be produced via a regulated process. For example, the PDE4DIP locus encoding phosphodiesterase 4D interacting protein shows marked tissue specificity for several AS transcripts with alternative first exons (Figure 5).
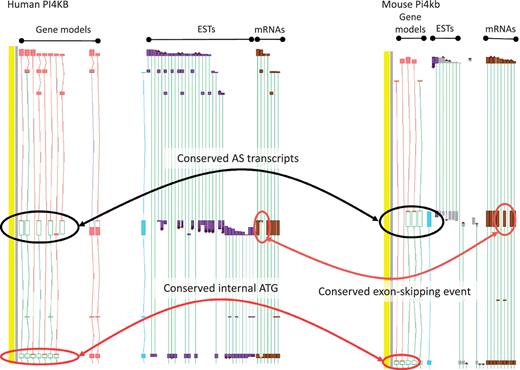
Zmap screenshot of the PI4KB/Pi4kb genes encoding phosphatidylinositol 4-kinase, catalytic, beta in human (left) and mouse (right) respectively. Black ovals highlight exon-skipping event observed in both species, orange ovals highlight the species-specific mRNA evidence supporting the annotation of the exon-skip event and red ovals highlight the conserved internal ATG likely to be utilised in the event of the exon-skip.
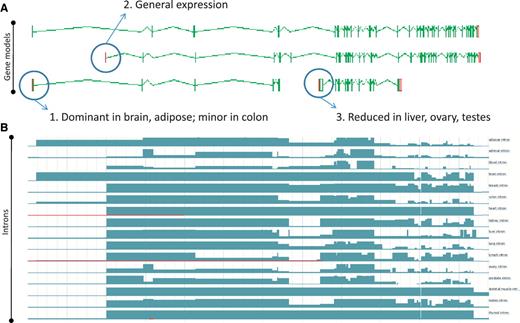
Zmap screenshot of four representative full-length transcripts at the human PDE4DIP locus encoding phosphodiesterase 4D interacting protein (A) and screenshot of the Ensembl display of introns built using the Illumia Bodymap2 RNA-Seq dataset (B). The three main transcription start sites (TSS) associated with the locus are circled and the primary tissue types in which the TSSs are up or downregulated are indicated.
AS and functional context
A full understanding of the AS transcript complement of a coding locus is self-evidently desirable in its own right. Furthermore, it can profoundly alter our interpretation of how changes to a locus, either via natural variation or experimental design can affect its function. For example, we recently investigated 800 variants (SNPs and small indels) that were predicted to lead to loss of function (LoF) of the loci in which they were found (22). Following full manual annotation of the loci and analysis of the impact of the LoF variants, ∼36% were shown to affect exons subject to AS. Clearly this could have a significant bearing on the functional potential of the affected loci. For example, an AS transcript that skips an exon may lead to the exclusion of the LoF site from the mature mRNA, avoiding any effect on function. Such a novel exon-skipping transcript could putatively ‘rescue’ the functional potential of a locus by encoding a CDS with some degree of functional complementation. Also, where the AS is tissue specific, any tissue that exclusively transcribes the exon-skipping form is likely to avoid any functional consequences of the LoF variant. Figure 6 shows the potential complementation for the loss of one variant of the PDE4DIP locus.
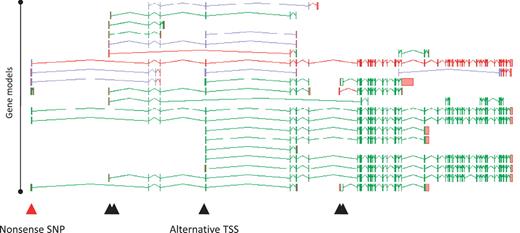
Zmap screenshot of all transcripts at the human PDE4DIP locus encoding phosphodiesterase 4D interacting protein. The red arrowhead indicates the position of a disabling nonsense SNP, black arrowheads indicate the positions of alternative TSSs.
An understanding of AS has also proven important for the production of large-scale knockout mouse resources, such as those generated by the EUCOMM and KOMP programs (50). Using a ‘knockout-first’ approach (51), the identification of a critical exon or exons common to all coding transcripts is essential for ablating gene function. Targeted exons are typically asymmetric, which when deleted induce a frameshift resulting in a truncated CDS that renders the transcript susceptible to NMD. In the absence of detailed manually annotated mouse gene structures, target exons were selected using automated gene models following an assessment of available transcriptome data (ESTs and mRNA). Subsequent validation of knockout designs by means of full manual annotation has revealed the complex implications of AS for targeted transcripts. For example in the Gls2 gene, initial targeting of exons 2–4 was shown to rescue a transcript previously susceptible to NMD that would result in a full-length protein being produced (Figure 7). Although the resulting protein would have a disrupted glutaminase domain, intact C-terminal Ank2 domains containing ankyrin-repeats would remain and may facilitate protein-protein interactions. Such a knockout strategy, targeting exons 2–4, therefore compromises a key criterion of the knockout programs to disrupt >50% of the wild-type protein.
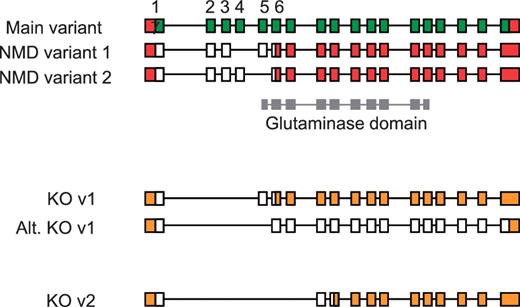
Validation of vector designs for Gls2 gene. The main exon structure of Gls2 (main variant) is displayed alongside NMD variants 1 and 2 based on mRNAs BC02566.1 and AK039618.1, respectively. Hypothetical knockout (KO) transcripts based on annotated structures are shown. Knockout transcript, KO v1, is derived from the main variant and NMD variant 1, where as the alternative version (Alt. KO v1) is derived from NMD variant 2. Exons 1 to 7 are numbered with coding regions displayed in green (main variant) or white (NMD and KO variants) and untranslated regions (UTR) shown in red or orange. The Gls2 glutaminase domain is shown in grey, overlapping exon structure.
Conclusions
Since manual annotation was initiated as part of our contribution to the human genome project we have adopted a high-sensitivity, high-specificity approach. Our identification and annotation of all experimentally supported transcript models leads to an enrichment of AS transcript present in our geneset compared to those found in other manually curated genesets. The structure of every model is manually checked, and we attempt to keep our annotation up to date with current science (for example systematically incorporating the annotation of NMD biotypes). We continue to be cautious in terms of assigning protein-coding potential in order to avoid making unsupported decisions that may result in contamination in other databases. This approach differs from other significant databases whose main aim is to provide full-length protein annotation. This approach has proved useful in capturing features that had no obvious function at the time of their annotation but have, in time, become better understood, for example lncRNA loci. Our annotation of AS transcripts lacking obvious function has also proved vitally important for example in the reclassification of variants identified by the 1000 Genomes project as causing LoF in human genes, and in mouse knockout projects. As our catalogue of AS events continues to expand, we will incorporate new methods to validate the functional potential of these transcripts through the use of data from new sequencing and proteomics technologies.
Funding
This work was supported by the Wellcome Trust [WT077198]; the National Human Genome Research Institute, National Institutes of Health, USA [5U54HG004555-04S1]; and the European Union Sixth Framework Programme EUCOMM (European Conditional Mouse Mutagenesis). Funding for open access charge: Wellcome Trust [WT077198].
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}