





Abstract
The PRoteomics IDEntifications (PRIDE) database is a large public proteomics data repository, containing over 270 million mass spectra (by November 2011). PRIDE is an archival database, providing the proteomics data supporting specific scientific publications in a computationally accessible manner. While PRIDE faces rapid increases in data deposition size as well as number of depositions, the major challenge is to ensure a high quality of data depositions in the context of highly diverse proteomics work flows and data representations. Here, we describe the PRIDE curation pipeline and its practical application in quality control of complex data depositions.
Database URL:http://www.ebi.ac.uk/pride/.
Introduction
Proteomics can be defined as ‘the study of the subsets of proteins present in different parts of the organism and how they change with time and varying conditions’ (1). While the correlation between gene expression and protein abundance in a given cellular system is a topic of ongoing scientific discussion, proteomics undoubtedly provides a unique means to study the biologically essential state of a protein, including post-translational modifications (PTMs), such as phosphorylation or glycosylation, essential for the modulation of protein activity, translocation and complex formation. Tandem mass spectrometry (MS) is the most commonly used technology for obtaining high-throughput proteomics information. Dramatic improvements in MS instrumentation and experimental approaches have allowed proteomics to move from the generation of simple protein lists for a given system to targeted observations of quantitative and dynamic proteome changes.
From a data management perspective, these changes have resulted in a rapid increase in the size and complexity of MS data sets supporting a particular scientific publication. Increased instrument precision and measurement frequencies result in much larger raw data sets, and improved experimental technologies now allow multiplexed observation of several proteome states, with the associated, which are much more complex descriptions of metadata and results. Figure 1 shows the top 20% largest PRoteomics IDEntifications (PRIDE) submission file sizes and spectra counts per submission over time, indicating at least one order of magnitude increase in file sizes and about two orders of magnitude increase in spectrum counts from 2006 to 2011. Figure 2 shows the overall growth of number of spectra, protein and peptide identifications over the same period.
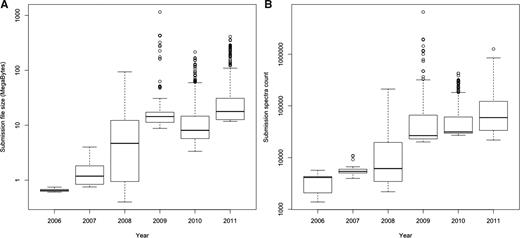
(A) Top 20% largest submission file sizes and (B) top 20% highest spectra count per submission file over time. The top 20% percentage of the submitted files is shown in order to reflect to the state of the art methodology and MS machines applied. The figure shows that there was at least one order of magnitude increase in file sizes and about two orders of magnitude increase in spectra counts from 2006 to 2011.
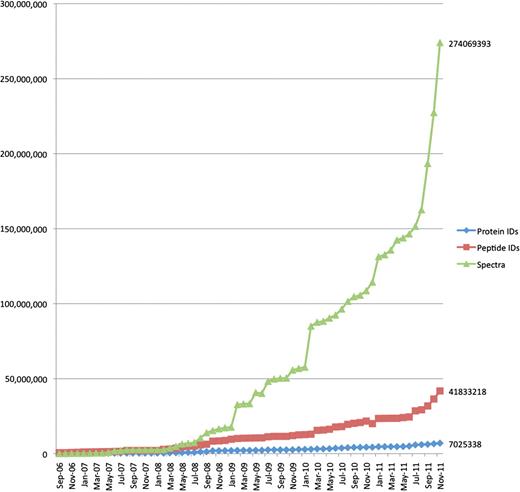
The overall growth of peptide and protein identifications, and mass spectra at PRIDE over time. The increase in data content of three core types of information stored at PRIDE: peptide and protein identifications and mass spectra, from 2006 to November 2011.
From a data management point of view, the hardest problem for proteomics repositories like PRIDE is presented by the many existing proteomics workflows. PRIDE is centered around the so called bottom-up proteomics approach, where the detected analytes are not complete proteins but peptides generated by enzymatic cleavage of the parent protein(s). There might be a separation step applied, like gel electrophoresis or, as in the so called shotgun proteomics methodology, the whole-protein extract is digested followed by the separation of peptides by liquid chromatography. The choice of workflows is much larger than in, for example, genomics or transcriptomics, a consolidation process focusing on a few workflows and vendors has not yet taken place. The experimental approach, the data processing workflow, and the type of data generated all influence the data formats that must be accepted by the proteomics repository. Each additional workflow provides an additional challenge to the unified representation of proteomics data in a single, structured repository.
Fortunately, the situation is already improving significantly as a result of the Human Proteome Organization Proteomics Standards Initiative (PSI) (http://www.psidev.info) developing the standard formats mzML (for MS data) (2) and mzIdentML (for protein and peptide identifications coming from MS experiments) (3), which are becoming increasingly implemented by instrument and search engine producers.
In a field where complex data sets containing hundreds of thousands of spectra, generated under multiple conditions, support scientific conclusions at the molecular level, the sufficient reporting of the experimental metadata is essential for quality assessment of these conclusions, as well as for a potential reanalysis of the valuable data. In an effort to support the systematic reporting of metadata, the PSI has developed a series of modular MIAPE (Minimal Information About a Proteomics Experiment) guideline documents (4), which state the desirable minimal information that should be reported per type of experiment. In parallel, proteomics journals have developed guidelines to ensure high-quality data and experimental approaches (5).
In addition to the increase of size and complexity of the individual data deposition, the number of data deposition requests to PRIDE has rapidly increased, from ‘occasional’ to a current average of two per working day, a trend we expect to continue. One reason is that key journals in the field, like Proteomics and Molecular and Cellular Proteomics, are increasingly mandating public deposition of MS data to support the publication of related proteomics manuscripts. At the same time, as a way to maximize the value of the funds provided, several funding agencies (e.g. the NIH in the USA, the BBSRC and the Wellcome Trust in the UK) are increasingly mandating the public availability of the produced experimental data.
While PRIDE is not the only large public proteomics data repository, its specific mission is the structured, searchable representation of proteomics data sets, as they support specific scientific publications based on direct data depositions by the data producers. Once a data set has been released, it will remain fundamentally unchanged, apart from maintenance tasks like updates to protein names imported from e.g. UniProtKB (6). This archival function is similar to the archival function of e.g. the EMBL/GenBank/DDBJ DNA repositories of the INSDC (7) and the recently closed NCBI Peptidome (8). In contrast, PeptideAtlas (9) and the Global Proteome Machine Database (GPMDB) (10) (re-) process proteomics data in their own processing pipelines, while Tranche (http://tranche.proteomecommons.org) can be compared to a giant hard drive, which provides a file-based repository of proteomics raw data with minimal metadata annotation.
The PRIDE curation pipeline
The PRIDE database (http://www.ebi.ac.uk/pride) was established at the European Bioinformatics Institute (EBI, Cambridge, UK) in 2004 as a public data repository to support the publication of MS-related studies. There are three main kinds of information stored at PRIDE: peptide and protein identifications and quantitation values, the corresponding mass spectra (the primary data used to infer the identifications) and as a key point, as much associated metadata as possible. The metadata, which are administrative (contact), technical (data processing, software used) and biological (sample), are captured by using controlled vocabulary terms, to support systematic search across data sets.
The PRIDE submission workflow is summarized in Figure 3. Due to the difficulty caused by the existing high heterogeneity in data formats, the PRIDE Converter application (http://code.google.com/p/pride-converter) was developed for streamlining data submissions to PRIDE (11). PRIDE Converter is an open source, platform-independent software tool written in Java. Data providers can use this tool to transform a large variety of popular MS proteomics formats into PRIDE XML (the internal PRIDE data format) via a graphical user interface. PRIDE Converter makes the submission of MS data a much easier and more straightforward process, especially for researchers without bioinformatics support. Supported file formats, data requirements and how to perform a submission are documented at http://www.ebi.ac.uk/pride/submissionGuidelines. The PRIDE Converter has definitely been the key factor in the huge growth in data contents in PRIDE since 2008. A reimplementation of this tool with extended functionality is currently under development and will be available in the first half of 2012.
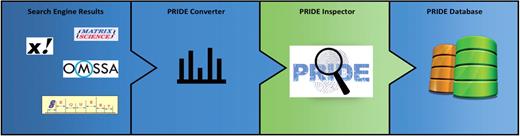
The PRIDE submission workflow. Search engine results containing identifications and spectra files are converted into PRIDE XML files by PRIDE Converter. Initial assessment and visualization of the data are done with PRIDE Inspector. This part is highlighted because that is where the bulk of the curation is happening. Finally the files are submitted to the PRIDE database.
At the beginning of 2011, we introduced a second tool called PRIDE Inspector (http://code.google.com/p/pride-toolsuite/), as a new open source Java application for visualizing and performing an initial assessment of MS data (12). PRIDE Inspector provides different views, each focusing on a different aspect of the data: experimental details, spectrum, protein, peptide, quantification values (if available) and summary charts. A major strength of PRIDE Inspector lies in its ability to perform an initial assessment of data quality, since a variety of simple charts based on the data are generated automatically. The PRIDE Inspector charts have been described in detail previously (12). With PRIDE Inspector, researchers can examine their own data sets before the actual submission to PRIDE is performed, or access data already in PRIDE for data mining purposes. It can also be used by journal editors and reviewers for the thorough review of submitted and private data at the pre-publication stage. And naturally, it can be used for the PRIDE curation tasks and basic quality checks.
Once the data have been successfully submitted to PRIDE, accession numbers are provided to the data submitter, as well as a ‘reviewer’ login and password. Via the journal editor, this can be provided to the manuscript reviewers, who can access the data in its final database representation in PRIDE. On publication of the manuscript, the supporting PRIDE data set is publicly released.
In our experience, significant errors can creep into the representation of complex proteomics data sets during the data deposition process, due to problems in our data deposition software and its representation of complex workflows, due to oversights by the data depositor, and sometimes because the data were pre-processed by a proteomics core facility or company, turning the whole process into a ‘black box’ for the researchers who are publishing the manuscript and therefore making the submission to PRIDE. Direct interaction between the PRIDE submission/curation team and the users then becomes critical to try to address these issues where possible.
Frequent data quality issues
Once the PRIDE XML files have been uploaded via FTP into PRIDE, the first thing needed is to check the syntax. It could be not compliant due to, for instance, missing or truncated XML tags. If that is the case, the file cannot be submitted. So, a XML syntactic validation checker is run against those files. This step is performed automatically by PRIDE Converter, so it is only actually done for files that have been generated by other pipelines.
Once the files have passed syntactic validation, the different type of quality issues generally fall into one of the following two categories:
(1) Core or metadata are missing. Two different scenarios are possible: (i) technically, it is feasible to provide the data or (ii) because the concrete proteomics workflow and related file formats are not supported by PRIDE Converter, the information cannot be easily provided.
A frequent example of missing core data is when peptide assignments and protein identifications are not uploaded, but only spectra information is provided. This can happen if the submitters ignored adding the search engine result files when converting the data into PRIDE XML files (for instance, they have only used mgf files to generate the PRIDE XML files instead of Mascot DAT files).
It can also happen that a non-supported search engine or additional post-processing software was used (for instance, at present the software Proteome Discoverer, from Thermo). So while it is not necessary to upload peptide assignments and protein identifications, we strongly recommend providing them. Additionally, the lack of identifications might even affect the outcome of the peer review process.
Another case of missing data is when the species information is not provided. Also it is frequent not to provide a project name or incomplete submitter details in case of third-party PRIDE XML export tools (like, for instance, the PLGS software from Waters).
(2) Inconsistent/incorrect data are uploaded.
The most frequent source of incorrect/inconsistent data is caused by the erroneous annotation of protein modifications, something that will be discussed in the next section.
PRIDE curation snippets
As an illustration of PRIDE curation, we are going to discuss the checks that are done with regard to the correct reporting of protein modifications can be natural (e.g. PTMs like phosphorylation or glycosylation) or artificial (e.g. cysteine alkylation introduced during sample preparation). Protein modifications can occur on all the 20 natural amino acid residues and on the amino and carboxy terminal position of the protein. Depending on the structure and position of the amino acid in the protein, there can be more than one modification on the same amino acid residue. A modification introduces a mass shift (also known as delta mass) in the peptide and in the resulting fragment ion and this is then detected by the search engine and then assigned to protein modifications.
Modification information is captured in PRIDE using the PSI-MOD (Modifications) ontology (13). The PSI-MOD ontology contains detailed information about each protein modification, such as delta mass, and amino acid specificity. It is important to highlight that tracking and annotating modifications is not a simple process. There are new modifications described on a regular basis, as this is an active field of research. As a result, potential new modifications need to be added to the PSI-MOD ontology before the data submission to PRIDE is done. For instance, a new modification named l-cysteine bacillithiol disulfide (MOD:01860) was introduced to PSI-MOD due to the submission of the already publicly available data set ‘Bacillithiolation and hypochlorite resistance’ (experiment accession numbers 17516–17659) (14).
Modifications can often be misreported or missing, and PRIDE Inspector provides built-in tools to address these issues. PRIDE Inspector calculates the ‘delta m/z’ values for the reported identified peptides by calculating the difference between the experimentally detected m/z value (corresponding to the precursor peptide ion m/z) and the theoretical mass of the peptide identified. If the resulting value is outside of a normal range (depending on the accuracy of the mass spectrometer used), this constitutes a good indication that something has gone wrong while annotating the data. For instance, an outlier value can indicate whether the precursor charge was wrongly assigned, or the protein modifications were not reported correctly. In the PRIDE Inspector ‘Peptide View’, the delta m/z values are displayed. Currently, the delta m/z values outside of the −4.0 to +4.0 m/z units range are highlighted in red, while the normal values are highlighted in green.
If the distribution of all the ‘delta m/z’ from the whole experiment (MS run) is taken into account, it can give a clear indication that something has gone wrong in the experimental set up, or that there has been a mistake in the reporting of the final results at a global level. This chart is available in the ‘Summary charts’ view in PRIDE Inspector (Figure 4).
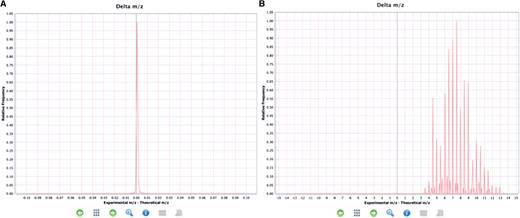
Examples of expected and unexpected delta m/z value distributions. (A) An expected delta m/z distribution where the value are within the −4.0, +4.0 m/z units range. (B) Unexpected distribution of delta m/z values where most of the values are outside of the −4.0, +4.0 m/z units range indicating a potential problem.
The following two examples (based on real data) demonstrate how protein modification-related issues were detected and solved during the submission process of different PRIDE data sets. In one of the examples provided, a protein modification was misreported, and in the other case a modification was omitted entirely.
Example 1: Misreported modification.
The submitter picked l-tyrosine residue (term MOD:00028 in the PSI-MOD ontology) as the reported protein modification. This protein modification converts a source amino acid residue to l-tyrosine, which does not cause a defined and concrete delta mass shift in the m/z value of the fragment ion that contains that tyrosine. The delta mass will be different for each amino acid that is substituted by l-tyrosine. This is why the delta mass information is missing from PSI-MOD.
In one file, an MS/MS spectrum was used to identify the peptide PVRYIR and the unexpected delta m/z value reported by Inspector was 39.98 Da (Figure 5A). Since the precursor charge assigned is +2, the approximate mass of the potential modification can be found by simply multiplying 39.98 by 2, yielding 79.96 Da as monoisotopic mass delta. Indeed, when informed about the PRIDE Inspector output, the submitter chose to use the MOD:00048 term, named O4′-phospho-l-tyrosine with a monoisotopic mass delta of 79.96 Da instead of the wrongly picked l-tyrosine residue. With a custom script, the modifications were replaced in the PRIDE XML file and the resulting file was checked with PRIDE Inspector (Figure 5B).
Example 2: Missing modification.
The PRIDE XML files prepared for submission did not contain originally an N-terminal modification for its 97 identified peptides, possibly due to a parsing problem. Depending on the charge state, this meant a systematic 44 delta m/z units for doubly charged, and 29.3 for triply charged precursor ions suggesting a mass of around 88 Da for the modification [(44 × 2) ∼ (29.3 × 3)] (Figure 6A). Generally, if the subtraction yielding a delta m/z value is positive, it means that something was not calculated in the theoretical m/z so the experimental m/z is bigger. Thioacylation (MOD:01223, name: thioacylation of primary amines - site N-term xref. DiffMono: ‘87.998286’) was added to all the peptides via custom script (Figure 6B).
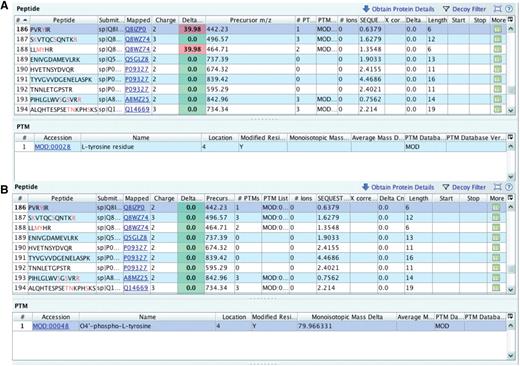
Checking delta m/z values with PRIDE Inspector peptide view: Example 1: misreported modification (A) outlier delta m/z value highlighted in red indicates a potential problem with the assigned protein modification. (B) protein modification replaced with the proper PSI-MOD term with a delta mass that gives an expected delta m/z value.
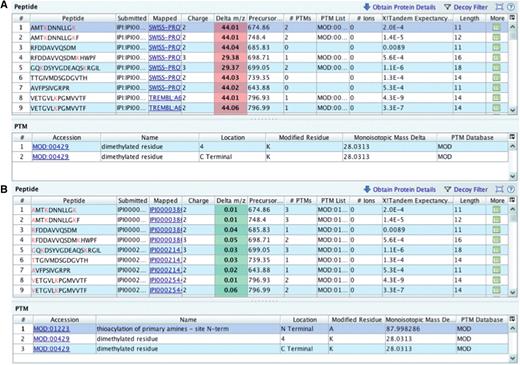
Checking delta m/z values with PRIDE Inspector peptide view: Example 2: missing modification (A) systematic outlier delta m/z values highlighted in red indicates a potential problem due to the lack of a fixed protein modification. (B) A PSI-MOD term was added that gives the expected delta m/z values for all peptides.
Conclusion
While generation and public availability of proteomics data are still, several orders of magnitude smaller than e.g. genomics data, both quantity and complexity of proteomics data sets deposited in the PRIDE database are rapidly increasing. Meanwhile, the complexity of proteomics data makes a fully automated data deposition process almost impossible. Curators play a very active role in supporting the data submitter in the preparation and quality control of a PRIDE data deposition. To cope with the workload of increasing, and increasingly complex data depositions, we have developed two key tools: the PRIDE Converter for preparation of a data deposition, and the PRIDE Inspector for initial assessment of a data set in PRIDE format. The PRIDE Inspector is offered as a tool to all key participants in the data publication process, namely the data generator/submitter, the curator and the manuscript reviewer appointed by the journal, implementing a multistage quality control process for data eventually published in support of a scientific publication.
At the current stage, automated enforcement of metadata annotation guidelines defined by journals or the PSI is not yet implemented, but we are in the process of developing increasingly complete validation procedures that identify and indicate missing or potentially erroneous elements of the deposited data set, supporting the community in the continued strive to increase the quality of publicly available proteomics data.
Funding
Wellcome Trust (grant number WT085949MA to The PRIDE database); EMBL core funding; EU FP7 LipidomicNet (grant number 202272 to J.A.V.); ProteomeXchange (grant number 260558). Funding for open access charge: Wellcome Trust (grant number WT085949MA to The PRIDE database).
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}