





Abstract
Genome-wide association studies (GWAS) query the entire genome in a hypothesis-free, unbiased manner. Since they have the potential for identifying novel genetic variants, they have become a very popular approach to the investigation of complex diseases. Nonetheless, since the success of the GWAS approach varies widely, the identification of genetic variants for complex diseases remains a difficult problem. We developed a novel bioinformatics approach to identify the nominal genetic variants associated with complex diseases. To test the feasibility of our approach, we developed a web-based aggregation tool to organize the genes, genetic variations and pathways involved in preterm birth. We used semantic data mining to extract all published articles related to preterm birth. All articles were reviewed by a team of curators. Genes identified from public databases and archives of expression arrays were aggregated with genes curated from the literature. Pathway analysis was used to impute genes from pathways identified in the curations. The curated articles and collected genetic information form a unique resource for investigators interested in preterm birth. The Database for Preterm Birth exemplifies an approach that is generalizable to other disorders for which there is evidence of significant genetic contributions.
Database URL:http://ptbdb.cs.brown.edu/dbPTBv1.php
Introduction
The promises of the genomic era have been presented eloquently (1–3). While it is clear that ‘genomic medicine’ is in its infancy, an impact on a number of important diseases and insights into the pathobiology of others have already been identified (1–3). Included among these is the recognition that minor variations in many different genes can form the basis for variation in disease susceptibility. They are also the substrate on which gene–environment interactions can occur. However, the promise of the genome era has also been met with skepticism as some results have been mixed (4–9). The genome-wide association study (GWAS) approach queries the genome in a hypothesis-free, unbiased approach, with the potential for identifying novel genetic variants. While there have been a number of important ‘hits’, for example, macular degeneration, inflammatory bowel disease, obesity (10–12), there are many ‘misses’ and failures to replicate findings even from large-scale studies. Moreover, a GWAS-based interrogation of large numbers of anonymous single nucleotide polymorphisms (SNPs) or copy number variations (CNVs) severely limits power and makes it nearly impossible, computationally, to examine combinatorial gene–gene interactions (13–15). However, employing pathway analysis or other a priori biological knowledge bases improves success in extraction of valuable information from GWAS analyses (16,17).
We are interested in the genetic architecture of preterm birth. We have developed an approach to identify a more manageable set of candidate genes, which nonetheless incorporates some elements of genome-wide investigation for the study of preterm birth. Our approach combines information from published literature with data from expression databases, linkage data and pathway analyses to identify biologically relevant genes for testing in an association study of genetic variants and preterm birth. We have developed a web-based, semantic data mining and aggregation tool to ‘filter’ published literature for evidence of association of preterm birth with genes, genetic variants, SNPs or changes in gene expression. A trained curation team extracted gene and protein information from published articles specific to preterm birth. Identified genes or sets of genes have been deposited into the database with reference PubMed Identifier (PMID) number and related information extracted from several resources (18–20). In addition, genes identified from archives of expression arrays and genomic regions identified from linkage analyses have been aggregated with the genes curated from the literature. Lastly, pathway analysis was used to impute genes from pathways identified during curation. These genes, their genomic location, the SNPs contained therein and any associated CNVs are presented in a searchable database.
The Database for Preterm Birth (dbPTB) is a robust resource for the community of biologists, perinatologists, geneticists and other investigators interested in the etiology of preterm birth or related phenotypes. Moreover, we believe this approach is generalizable to investigation of other disorders where there is evidence for important genetic contributions. The resources supporting this approach have been made available in a publicly accessible database at http://ptbdb.cs.brown.edu/dbPTBv1.php.
Methods
Retrieval of data and updates
The Database for Preterm Birth (dbPTB) was implemented using a MySQL database running on a Linux server with PERL and PHP scripts used for all data retrieval and output. dbPTB used SciMiner™ to extract the gene and protein information from published articles specific to preterm birth (21). From the 18 million records representing 22 000 journals that are housed in PubMed, we used computational data mining to extract more than 30 000 articles related to preterm birth and potentially including relevant information on genes, SNPs or genetic variations. From further refinements of the semantic language processing, we identified 981 articles with putative information about genes and genetic variants associated with preterm birth. For the retrieval of articles to be curated, we used several different approaches. First, we used queries which have common and very well known keywords for preterm birth and genetics, e.g. ‘preterm birth and genes’. Second, after acceptance of extracted articles, we annotated all the medical subject heading (MeSH) terms associated with these papers. These were used to create new search queries incorporating the newly annotated MeSH terms. We called these two approaches ‘forward and reverse curation’. Third, the reference lists of each article under curation were also carefully examined and potentially relevant articles were extracted through SciMiner™ for curation. Bimonthly search-runs for articles for curation are used to update the database regularly.
Curation
All the filtered articles putatively contain information on genes, gene–gene interactions and SNP information related to preterm birth. To evaluate this evidence, we created a curation team to read each publication. The team consisted of researchers and medical students formally trained in the molecular and cell biology and genetics of preterm birth. Each article was carefully read. Attention was devoted in particular to study design, relevance of the article to preterm birth per se and not issues related to prematurity but distinct from preterm delivery. Articles that contained relevant, statistically documented information on genes or genetic variants related to preterm birth were ‘accepted’ and deposited into the database with their unique PMID. Also entered into the database from each article were the genes, genetic variants, SNPs, RefSNP accession ID (rs number) (when available) and annotations describing gene–gene interactions shown to be statistically significantly related to an increased risk for preterm birth. We accepted in all cases the authors’ criteria for statistical significance. All genes and genetic variants entered into the database were entered using their unique HGNC numbers for identification. SNPs were entered into the database and recorded with their appropriate rs number using HapMap Data Release 27 (22). Where specific haplotypes were shown to confer significant risk for preterm birth, all the individual SNPs within the haplotype were entered into the database. This was true even if by univariate analysis an individual SNP was not statistically associated with increased risk for preterm birth. Since they represent significant confounding factors in the risk and pathogenesis of preterm birth, the association of premature rupture of the amniotic membranes (PROM) and/or evidence of intra-amniotic infection with preterm birth were recorded. Thus, their association with preterm birth individually is searchable within the database. Lastly, for curation, in a minority of articles, animal models rather than results from human patients were reviewed. Similar criteria were used for ‘acceptance’ and inclusion of genes. In the case of data from mouse, rats or other species, the human homolog was entered into the database, again by its unique HGNC number.
Inter-rater reliability was assessed and κ scores were measured after training (23, 24). Inter-rater reliability was maintained by formal, weekly ‘curation meetings’ where difficult publications, or any publication a curation team member felt would be useful for discussion and comparison, were reviewed conjointly. We designed and built a separate database for the curation process, which allowed remote login, password protected access to full text of the articles via the Brown University Library eJournals collection. This allowed annotation of the articles, putative genes, SNPs and variants contained in the extracted papers. Since the curation database allowed curators to work remotely, it significantly accelerated the process of curation. Articles which are accepted for preterm birth immediately become accessible to dbPTB queries along with all the relevant genetic data (Figure 1). An algorithmic description of the curation process in detail is shown in Supplementary files.
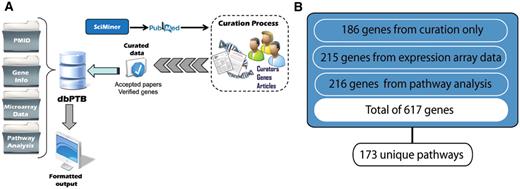
(A) Workflow for retrieval of articles, curation and extraction of genes from literature, microarray data and gene interpolation for pathway analysis. (B) Total number of genes, their associated original sources and number of unique pathways represented.
Database queries
Voluntary practices by many investigators and the development of mandatory data sharing policies for federally funded projects have made available collections of high dimension databases of expression data, data from linkage analyses, databases of results from SNP arrays and data from proteomic platforms. This includes transcriptome wide data comparing RNA levels from tissues from preterm deliveries with similar samples from term delivery. The database queries may also include genomic regions identified from linkage analyses and the SNPs and genes therein. These resources were searched for genes, genetic variants and proteins related to preterm birth or showing differential association with preterm birth. We searched publically available databases and, likewise, articles describing genome- or transcriptome-wide analyses. We also searched for articles that provided information on analyses of proteins in body fluids or compartments that were analyzed using contemporary proteomic techniques like mass spectrometry. Lastly, we searched new repositories from the Heart, Lung, Blood Institute and the National Human Genome research (NHGRI), including the Human Gene Mutation Database and the Catalogue of Published Genome-Wide Association Studies hosted by the NHGRI. From databases or articles on transcriptome-wide analyses, we again used the individual authors’ criteria for statistical significance. We included genes whose expression was statistically increased or decreased in association with preterm delivery. Likewise, for proteomic analyses, we included genes and proteins whose unusual presence in a body fluid suggested a possible relationship to the pathophysiology of preterm birth, e.g. proteomic analysis of amniotic fluid.
SNP data
SNP data for each of the genes included in dbPTB is also included in the database. The first source of this information was from the literature curation itself. Wherever noted by the original authors, we included specific SNPs (by rs number). We also included specific polymorphisms for which there was published information. The second and larger source of SNP data in dbPTB comes from HapMap. We include all the tag SNPs for each gene from HapMap release number 27. The nominal haplotype block size in from the HapMap investigations is 2–10 kb (22), so we included all tag SNPs from 5-kb upstream to 5-kb downstream from the genomic sequence.
Data integration
As noted earlier, during the curation process, if an article supported a specific gene, genetic variant, SNP or haplotype block, then those gene(s) and genetic variants were deposited into dbPTB with the reference article anchored by its unique PMID number. For each deposited gene, its related information and SNP data were gathered. Gene information was extracted from NCBI Entrez Gene and HGNC. NCBI dbSNP Build 126 was used for SNP information. We also collected all MeSH terms provided by the National Library of Medicine from the curated articles, which were accepted into the database. For each article, we also stored the abstract and related information such as title, journal and authors.
Pathway analysis
The Ingenuity Pathway Analysis (IPA, Ingenuity® Systems, www.ingenuity.com) tool was used to identify pathways and networks encompassing the genes we identified with significant evidence for their involvement in preterm birth. For this portion of the analysis, we used the genes which were retrieved during the literature search. We also included the genes and genetic variants identified in public databases, largely transcriptome wide array data sets (25, 26) and some proteomic analyses related to preterm birth (27). The genes identified by the Ingenuity pathway analysis were enterer into the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. This allowed us to identify the number and identity of pathways each gene or variant was associated with.
Results
Curation results
From 31 018 articles dealing with preterm birth extracted from PubMed by SciMiner, the ‘filtered set’ included 981 articles for which there was likely information about genes and genetic variants. These articles contained information on more than 1200 putatively related genes. From among these articles, with over 5000 associated the MeSH terms, we ‘accepted’ 142 articles described by a total of 960 unique MeSH terms. These articles contained statistically valid associations of 186 genes with preterm birth. The top 15 journals from which we extracted articles for curation are shown in Table 1. As can be seen, these were largely clinical specialty journals. Likewise, we identified and imported 215 genes from both published and public databases containing array data and data from other proteomic analyses. We included an additional 216 genes based on the interpolation from pathway analysis. These genes were contained in 173 unique pathways. A pathway diagram showing the workflow supporting retrieval of genes from the literature and public databases and gene interpolation from pathway analysis is shown in Figure 1.
Top 15 Journals with articles extracted for curation in dbPTB
| Journal | Number of articles for curation |
|---|---|
| 1. American Journal of Obstetrics and Gynecology | 84 |
| 2. Pediatric Research | 46 |
| 3. Pediatrics | 34 |
| 4. The Journal of Pediatrics | 32 |
| 5. Obstetrics and Gynecology | 17 |
| 6. Biology of the Neonate | 14 |
| 7 The Journal of Clinical Endocrinology and Metabolism | 13 |
| 8. Journal of Perinatology | 13 |
| 9. Journal of Perinatal Medicine | 13 |
| 10. Archives of Disease in Childhood Fetal and Neonatal Ed. | 12 |
| 11. Human Molecular Genetics | 12 |
| 12. International Journal of Gynecology and Obstetrics | 11 |
| 13. American Journal of Reproductive Immunology | 11 |
| 14. Proceedings of the National Academy of Sciences | 10 |
| 15. Endocrinology | 9 |
These results are all available in the Database for Preterm Birth (http://ptbdb.cs.brown.edu/dbPTBv1.php). Currently, the dbPTB contains 617 genes (186 from literature curation, 215 from microarray and proteomic databases and 216 from pathway interpolation). The specific origin of inclusion is retrievable from dbPTB and also shown in Supplementary Table S2. Also included in dbPTB are the 156 963 SNPs contained with the genomic and flanking regions of each gene in dbPTB. We have physically mapped the genomic location for genes in dbPTB. This will facilitate a number of investigations, including a more efficient approach to GWASs to investigate preterm birth and/or resequencing genomic regions with a more dense coalition of genomic variations. Figure 2 shows a diagram of all chromosomes and the number of genes mapped to each. As can be seen, there were no genes that we retrieved from the literature curation, databases or pathway analysis that mapped to the Y chromosome. Figure 3 shows a representative distribution of genes on chromosomes 6 and 11 as well as an expanded view which shows even greater resolution for a gene rich region on chromosome 11. Across the entire genome, there were genomic regions where the gene density was quite low with up to 60 Mb separating identified genes. There were also many regions with identified genes in close proximity with as little as 1 kb separating the genomic sequences. These results are provided in dbPTB and in Supplementary Table S3.
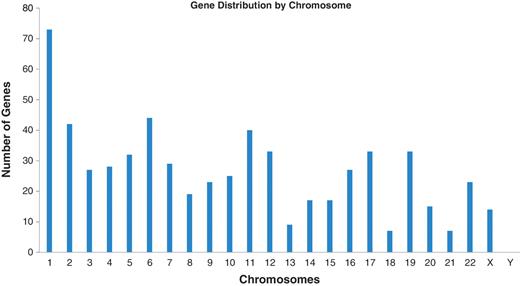
Number of genes among chromosomes identified from curated articles, databases and pathway analysis.
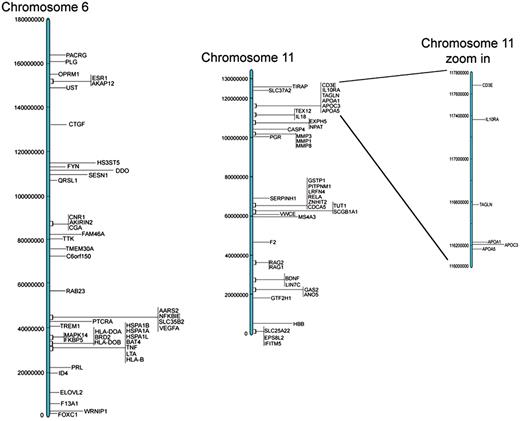
Representative examples of chromosomal location of genes for chromosomes 6 and 11.
Pathway information
A total of 25 networks were identified. The top functions described by pathway analysis are listed in Table 2. Among the major networks detected, several networks, ‘Inflammatory Response, Small Molecule Biochemistry, Cellular Development, Hematological System Development and Function, Cardiovascular Disease, Cellular Function and Maintenance, Connective Tissue Development and Function, Drug Metabolism, Genetic Disorder’ represented the largest portion of interaction domains.
Top functions of genes identified by pathway analysis
| Function | Number of networks |
|---|---|
| Inflammatory Response | 6 |
| Small Molecule Biochemistry | 5 |
| Cellular Development | 4 |
| Hematological System Development and Function | 4 |
| Cardiovascular Disease | 3 |
| Cellular Function and Maintenance | 3 |
| Connective Tissue Development and Function | 3 |
| Drug Metabolism | 3 |
| Genetic Disorder | 3 |
| Cell Signaling | 2 |
| Cellular Assembly and Organization | 2 |
| Connective Tissue Disorders | 2 |
| Embryonic Development | 2 |
| Hematological Disease | 2 |
| Infectious Disease | 2 |
| Inflammatory Disease | 2 |
| Lipid Metabolism | 2 |
| Molecular Transport | 2 |
| Amino Acid Metabolism | 1 |
| Antigen Presentation | 1 |
| Antimicrobial Response | 1 |
| Carbohydrate Metabolism | 1 |
| Cardiovascular System Development and Function | 1 |
| Cell Cycle | 1 |
| Cell Death | 1 |
| Cell-mediated Immune Response | 1 |
| Cell-To-Cell Signaling and Interaction | 1 |
| Cellular Compromise | 1 |
| Cellular Growth and Proliferation | 1 |
| Dermatological Diseases and Conditions | 1 |
| DNA Replication | 1 |
| Hematopoiesis | 1 |
| Infection Mechanism | 1 |
| Nucleic Acid Metabolism | 1 |
| Organismal Functions | 1 |
| Organismal Injury and Abnormalities | 1 |
| Organismal Survival | 1 |
| Organ Morphology | 1 |
| Recombination and Repair | 1 |
| Skeletal and Muscular Disorders | 1 |
| Skeletal and Muscular System Development and Function | 1 |
| Tissue Morphology | 1 |
The number of times each gene was included in different networks is also shown.
Database content and functionality
dbPTB allows several query strategies to search related articles, genes, SNPs, chromosomes or keywords against the MeSH terms and abstracts of the curated articles. If a user searches a gene of interest, and this gene is supported by articles in the database, the output will include all the articles supporting evidence for the queried gene's relationship to preterm birth. This includes the title of the articles, name of the published journal and the link to the original source, which most cases is NCBI PubMed. Moreover, information about the gene and related links are shown. This also includes links to Online Mendelian Inheritance in Man (OMIM), the UCSC Genome Bioinformatics and Hugo Gene Nomenclature (HGNC). Under the same search option, users are able to see all related SNP data for each gene. For each SNP, they are able to follow the link to the original source. They also have an option to download all rs numbers for the queried gene. In other searches, the users can get the genes for a specific chromosome and then again the related supporting evidence.
Discussion
We developed dbPTB, the database for preterm birth, to create a more manageable set of genes and genetic variants that may be involved in preterm delivery. We reasoned that this smaller set of candidates may allow important but otherwise difficult computational approaches to examination of gene/gene interactions in combinatorial or high-order fashion. We used the published literature as the first basis for population of this database. Web-based semantic data mining followed by careful manual curation was used to recover over 981 articles. These articles contained putatively nearly 1200 genes or genetic variants potentially related to preterm birth. We ‘accepted’ 186 genes out of this 1200. While literature curation provides access to the known information on genetic variants associated with preterm birth, it is not hypothesis-free. It is not a discovery-based approach. In order to add a discovery approach to our strategies, we also screened publically available databases for information on preterm birth. We reasoned that databases providing results from expression arrays or transcriptome-wide interrogations of tissues or body fluids comparing preterm deliveries with similar samples from those at full term would provide a hypothesis-free interrogation. We were equally interested in genes whose expression was either increased or decreased. We also searched for databases of proteomic results that might provide clues to preterm birth. The genes representing the combination of these search strategies were then entered into a pathway analysis. We used both Ingenuity and the KEGG pathway (28). Our interest was not to exclude all but those pathways with the greatest statistical validity. Rather, we sought to identify additional candidate genes who were clearly nested within important pathways represented by the genes retrieved by our search strategies, but whose only reason for exclusion was failure to be interrogated experimentally. We identified 186 genes using the literature-based curation, 215 genes from publically available databases and an additional 216 genes from the pathway-based interpolation. This total of 617 genes represents a parsimonious but robust set of genes for which there is good evidence for involvement in preterm birth. These genes and genetic variants can be used now in case–control studies comparing genetic variants, SNPs or CNVs. By physical mapping, these genes also point us toward candidates regions for efficient strategies for re-sequencing in search of rare variants. We believe this approach to be generalizable to other diseases and phenotypes.
GWASs have become a very contemporary and popular approach to the investigation of complex diseases (29). They have been made feasible through advances in technology and reduced costs (30). They have many great attractions; especially the prospect of discovery of new insights and novel gene–gene interactions not previously recognized (14–16). However, genome-wide approaches have also failed to demonstrate the ‘missing heritability’ in many common diseases (9, 31–34). There are several factors contributing to skepticism about the strength of this approach. First among these is that the majority of SNPs that have been identified through this approach are rarely the causative variants (9). At best, they are in linkage disequilibrium with the underlying pathogenic variant. Even more frustrating has been the modest if not exceptionally low effect sizes associated with the genetic variants that have been identified in most GWASs (6, 7). The low effect sizes suggest that the underlying pathophysiological causes, if they are genetic, are due to gene–gene interactions, gene environment interactions or other mechanisms. However, pair wise or higher order combinatorial effects from gene–gene interactions present difficult computational challenges with the large number of polymorphisms used in the majority of recent GWA studies (14). Importantly, new computational approaches have been developed which have identified gene–gene interactions in large data sets (13–16). In some cases, these approaches have been successful in identification of important genetic associations in studies which failed to identify main effects from individual variants (16,35–37). Moreover, when coupled to pathway based analysis or other approaches that use a priori biological knowledge, these newer computational approaches aid greatly in identification of important genetic contributions to risk in complex diseases (16).
The genetics of preterm birth and approaches to identify discrete genetic contributions to risk of preterm birth have been discussed (38–44). Recent studies have focused on genomic and proteomic approaches to diagnosing and determining the mechanism(s) of preterm labor. Polymorphic changes in the protein coding regions of specific genes and in regulatory and intronic sequences have been described. In most of the studies reported to date, candidate genes or proteins involved in inflammatory reactivity or uterine contractility have been investigated (34,38–55). Summaries of these observations and candidate genes have been reported (42). Most of the studies reported to date have involved modest sized patient cohorts and polymorphisms from genes involved in infection/inflammation. The results suggest that alteration in the structure and/or expression of these proteins interacts with infection and/or other environmental influences and is associated with preterm birth. The results generally, however, do not provide insight into the causes of prematurity in the absence of inflammation. They also do not demonstrate whether the observed associations are reflective of genetic mechanism(s) and/or gene–environmental interactions.
It is important to identify the strategies that have been used, the strengths and weaknesses of different approaches and recent, representative examples from the literature. Studies of the genetics of preterm birth are complicated by numerous confounders. These include: imprecise, non-uniform definitions; differences in the etiology of preterm delivery; the profound impact of environmental influences like PROM, inflammation, drug use or other significant clinical factors; the likely involvement of multiple loci and/or genes and complex patterns of inheritance. A precise estimate of the contribution(s) of genetic factors to preterm birth has been hard to achieve (38–44). Twin studies suggest heritability is up to 36%; however, differences in the definition of what constitutes a preterm delivery cloud the precision of those estimates (56, 57). The history of a previous preterm birth is one of the best predictors of recurrence of preterm birth. Likewise, the observation that mothers who were preterm or have a first-order relative with preterm birth are at increased risk of delivering prematurely both underscore the importance of genetic factors (40). Sisters are more likely to be concordant for preterm birth (16%) than sisters in law (9%) (58). A large study examining kinships in Utah identified closer genetic relationships among families with preterm birth than those without (59). The veracity of this observation is considered reasonable because the study was conducted among a population group with a lower incidence of some of the confounding environmental influences known to be associated with preterm birth (e.g. drug use and alcohol). Whether fetal or parental genes contribute to the risk of preterm birth has been investigated in several studies. One of the aforementioned twin studies which used birthweight in its ‘definition’ of prematurity noted maternal effects to account for 40% of the variance in birthweight and fetal factors to only account for 19% (60). This has been challenged, however, by a larger study suggesting 70% of the variance in birthweight is due to fetal genes (61). The majority of the studies suggest that paternal factors are less important in determining gestational length or birthweight (61, 62). More recently, large epidemiological studies drawn from population-based analyses in Sweden and Denmark support a predominantly maternal origin for the genetic contribution(s) to risk of preterm birth with little contribution by paternal or fetal genetic factors (63–66). Only one linkage analysis and analysis of quantitative trait loci to identify regions on specific chromosomes was ascertained because large pedigrees with a family history of preterm birth have been difficulty to acquire (67). Some discrete, single gene disorders, like the relationship of Ehlers Danlos syndrome to PROM and resultant preterm birth, have been identified (68). Thus, while there is sufficient information to suggest important genetic contribution(s) to the risk of preterm birth, the epidemiological evidence and pattern of inheritance all suggest that, similar to other complex diseases like hypertension, diabetes and some psychiatric disorders, preterm birth is a complex, polygenic disorder and likely entails activation and/or suppression of a host of genes (69).
Our approach is predicated on the notion that, if SNPs are contributing to the risk of preterm birth, they are likely to interact in more than a simple additive fashion. Therefore, a more manageable set of variants is needed in order to begin to address the computational power needed to identify those interactions. Our approach also allows physical mapping and demonstration of significant clustering of the genes associated with preterm birth across the genome. These carefully curated articles and collected genetic information form a unique resource for investigators interested in Preterm Birth.
Conclusion
The resource we have developed is useful because all the data associated with the disease of interest (SNPs, genes, variants and articles) are collated into a single source. The dynamic nature and query options of dbPTB enable user friendly access. The user interacts with dbPTB through a web interface specifically built with flexible searching capabilities and a robust output with supported links to original sources for people familiar with genetics and basic sciences as well as largely clinical scientists. We believe this approach is generalizable to other disorders for which there is evidence of significant genetic contributions. The generalizability of dbPTB to other diseases and phenotypes applies to not only the literature curation and database searching but also the pathway-based interpolations for probable candidates. Moreover, this approach may aid in identification of regions to search for rare variants and narrow the list of putative genes to a workable number so they can be assessed for their contribution to PTB in an experimental model. The resources supporting this approach have been made available into a publicly accessible database. The scripts and code are available from the authors on request.
Funding
National Foundation March of Dimes Prematurity Initiative (No. 21-FY08-563); National Institutes of Health (Grants NIH-5T35HL094308-02 and NIH-NCRR P20 RR018728).
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}