





Abstract
Recent advances in high-throughput DNA sequencing technologies have equipped biologists with a powerful new set of tools for advancing research goals. The resulting flood of sequence data has made it critically important to train the next generation of scientists to handle the inherent bioinformatic challenges. The North East Bioinformatics Collaborative (NEBC) is undertaking the genome sequencing and annotation of the little skate (Leucoraja erinacea) to promote advancement of bioinformatics infrastructure in our region, with an emphasis on practical education to create a critical mass of informatically savvy life scientists. In support of the Little Skate Genome Project, the NEBC members have developed several annotation workshops and jamborees to provide training in genome sequencing, annotation and analysis. Acting as a nexus for both curation activities and dissemination of project data, a project web portal, SkateBase (http://skatebase.org) has been developed. As a case study to illustrate effective coupling of community annotation with workforce development, we report the results of the Mitochondrial Genome Annotation Jamborees organized to annotate the first completely assembled element of the Little Skate Genome Project, as a culminating experience for participants from our three prior annotation workshops. We are applying the physical/virtual infrastructure and lessons learned from these activities to enhance and streamline the genome annotation workflow, as we look toward our continuing efforts for larger-scale functional and structural community annotation of the L. erinacea genome.
Introduction
The advent of next generation sequencing technologies has led to a dramatic change in the way many biologists approach their research questions and hypotheses. This shift toward such data intensive methodologies demands bioinformatics research infrastructure in the form of not only physical data connectivity and computational resources, but also the expertise and tools necessary to perform research that was until recently the province of genome centers and government initiatives.
Sequencing and annotation of the little skate (L. erinacea) genome is an ongoing project undertaken by the North East Bioinformatics Collaborative (NEBC)—a collaborative effort of the bioinformatics core facilities in the five NIH IDeA/NSF EPSCoR-supported states of the northeastern US [Delaware (DE), Maine (ME), New Hampshire (NH), Rhode Island (RI) and Vermont (VT)]. The NEBC was born out of the larger North East Cyberinfrastructure Consortium (NECC), a partnership that aims to build the critical infrastructure with physical resources and cyber-knowledgeable research scientists necessary to promote cutting-edge research within its member states, while leveraging complementary resources and expertise across the Consortium (http://www.necyberconsortium.org).
The Little Skate Genome Project (http://skatebase.org/) serves as a demonstration project for performing relevant collaborative research across the five states, while simultaneously developing the tools for data sharing and analysis that make such research possible. To introduce and expand bioinformatics expertise among life scientists in the region, Genome Annotation Workshops and Jamborees have been introduced as an integral part of this project, providing training in genome sequencing, annotation and analysis to researchers of all levels—students, postdoctoral fellows and junior faculty—including those from undergraduate and underrepresented institutions that do not have established research infrastructures. Following three 1-week-long workshops, a series of Mitochondrial Genome Annotation Jamborees were organized to coordinate the annotation of this first completely assembled element of the little skate genome. This article describes the educational, collaborative and scientific aspects of the Little Skate Genome Project with a focus on utilizing workshops and jamborees for promoting collaborative community annotation.
Little skate (L. erinacea) as a model organism
Little skate (L. erinacea) is one of 11 non-mammalian organisms selected for genome sequencing by an NIH National Human Genome Research Institute advisory panel because the skate shares characteristics with the human immune, circulatory and nervous systems. Leucoraja erinacea is a chondrichthyan (cartilaginous) fish native to the east coast of North America, ranging from North Carolina to Nova Scotia. As the most basal surviving clade of jawed vertebrates, chondrichthyans can provide unique insight into the origin and evolution of many developmental processes, at both the morphological and molecular level. Chondrichthyans exhibit many fundamental vertebrate characteristics, including a neural crest, jaws and teeth, an adaptive immune system and a pressurized circulatory system. These characteristics have been exploited to promote significant understanding about human physiology (1), immunology (2), stem cell biology (3–5), toxicology (6), neurobiology (7) and regeneration (8). For example, studies of hepatic homeostasis, detoxification and membrane transport using L. erinacea primary hepatocytes offer significant advantages over mammalian hepatocyte cultures as they retain hepatobiliary polarity for at least 8 h (9). The development of standardized experimental protocols in elasmobranchs such as L. erinacea and the dogfish shark (Squalus acanthias) has further positioned these organisms as important biomedical and developmental models. Despite this distinction, the only reported chondrichthyan genome is the low coverage (1.4×) draft genome of the elephant shark (Callorhinchus milii) (10).
To close the glaring evolutionary gaps in available elasmobranch genome sequence data, and concomitantly generate critical genomic resources for future biomedical study, the genome of L. erinacea was chosen over other elasmobranchs for a number of reasons. (i) At an estimated 3.42 billion base pairs across 49 chromosomes, the size of the L. erinacea haploid genome is approximately half that of the genomes of some other candidate elasmobranch model organisms such as S. acanthias (11), thus allowing better coverage from comparable sequencing efforts. (ii) Researchers at NECC member institutions have already generated a number of complementary resources that will facilitate the assembly and annotation of the skate genome including: embryonic transcriptome sequence (12) generated at Mount Desert Island Biological Laboratory (MDIBL) for Dr Randall D Dahn by the Centre for Applied Genomics at the Hospital for Sick Children (Toronto), a 4× coverage BAC library made for the Maine INBRE program by the Clemson University Genomics Institute, and approximately 31 000 Expressed Sequence Tags sequenced from three cDNA libraries (13). (iii) As the little skate is more experimentally tractable than the dogfish shark, the L. erinacea genomic sequence can be more readily leveraged.
As close evolutionary relatives, the little skate sequence will facilitate studies that employ dogfish shark and other elasmobranchs as model organisms. Furthermore, genomic sequence from L. erinacea will provide phylogenetically critical data of a chondrichthyan that is currently lacking for studies of molecular evolution and comparative genomics, ultimately advancing our understanding of basic human biology and disease.
Little Skate Genome Project overview
The NEBC employs a distributed model for the collaborative use of specialized resources and expertise in an integrated process for the Little Skate Genome Project (Figure 1), while maintaining an environment for active engagement of scientific leaders, including face-to-face meetings and weekly videoconferences between the participating groups.
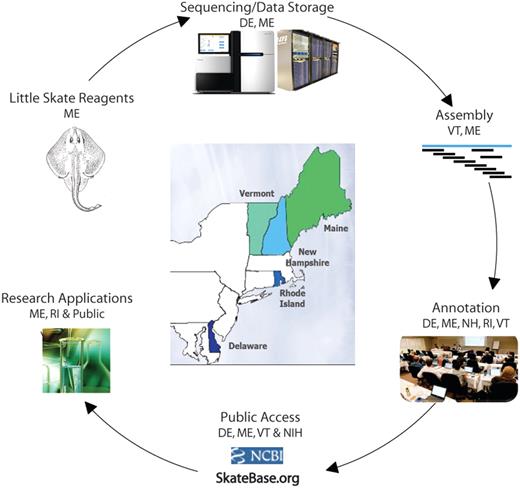
Little Skate Genome Project overview, illustrating the North East Cyberinfrastructure Consortium's distributed and collaborative resources.
The collaborative workflow (Figure 1) encompasses: (i) tissue sample collection and DNA extraction from MDIBL in Maine; (ii) DNA sequencing using Illumina HiSeq2000 and Genome Analyzer IIx at the Sequencing and Genotyping Center at the University of Delaware (UD); (iii) sharing of sequencing data among the partner institutions through the NECC Shared Data Center jointly housed by UD and the University of Maine (UM) using a suite of data sharing tools developed by the University of Vermont (UVM); (iv) assembly of sequence reads at UVM and MDIBL; (v) sequence annotation by participating groups from all five states using a bioinformatics analysis framework developed at UD; (vi) data dissemination via the SkateBase website and public repositories at the National Center for Biotechnology Information (NCBI); and (vii) ongoing utilization of the genomic data by the NEBC partners (ME, RI), as well as the larger elasmobranch research community.
Among the genomic contigs assembled to date, the mitochondrial genome sequence is the first completed element. To coordinate the annotation of the mitochondrial genome, a series of state-level Annotation Jamborees were held to generate draft annotations, which are then discussed and finalized in videoconferences attended by leading scientists from each state. The genome sequencing, assembly and annotation of the little skate chromosomes are still ongoing.
There are several layers of annotation that can be applied to a genome. The initial phase involves identification of genome features, such as open reading frames (ORFs), tRNA, rRNA, other non-coding RNA, regulatory sequence motifs, etc. Next automated annotation pipelines will apply tools such as BLAST and HMMER to identify putative features based on sequence homology to other better-characterized organisms. The manual curation that follows will verify, modify and/or expand upon automated annotations at both gene and protein levels to provide quality annotations for the genome. These initial annotations will continue to be refined as more experimental characterizations are published and data become publicly available. The collaborative processes will be further developed for completing the little skate genome sequencing and annotation project.
Collaborative tools
With the increased connectivity afforded by the NECC cyberinfrastructure, a number of shared computational resources and online tools were developed by UVM and UD to facilitate this collaborative project. When appropriate, pre-existing open-source software tools are adopted and customized to meet the needs of the project.
The SkateBase website (http://skatebase.org) is a central hub for the Little Skate Genome Project providing a platform for organizing, analyzing and disseminating information (Table 1). The SkateBase currently provides a number of tools that are accessible to internal curators and project personnel for file exchange, sequence analysis and collaborative annotation. The SkateBase File Exchange allows project members to share large next-generation sequencing files dynamically through an intuitive drag-and-drop web interface. The File Exchange tool utilizes the NECC Shared Data Center infrastructure to store, backup and transfer files. The SkateBase Community is a wiki-based tool to support the NEBC community annotation. The framework allows the creation of annotation templates that can be completed, reviewed and modified by curators, and serves as a clearing house to store annotations collected from annotation workshops and jamborees. Sequence analysis and visualization tools are used by curators to inform curation decisions. The tools include: (i) SkateBLAST, which provides homology search against an array of Skate-centric BLAST databases. SkateBLAST is derived from ViroBLAST (14), with a customized interface for job submissions to a UD high-performance computing cluster during heavy computational loads such as during Annotation Workshops. (ii) GBrowse (15) (http://gmod.org/wiki/GBrowse), which provides visualization of tracks of annotations completed and in progress; (iii) Mauve, a multiple genome alignment viewer (16, 17), and (iv) RACE-P (http://pir.georgetown.edu/pirwww/race_p/race_p_skate.shtml), which provides an interface for protein curation, including protein name, GO functional annotation, and sequence features such as signal peptide, domains and motifs. As the project progresses, many of the currently internally (NEBC curator-only) accessible tools and data will be made publicly available from SkateBase (Table 1), to promote expanded community annotation and data dissemination.
SkateBase components
| Component | Description | Public access |
|---|---|---|
| Informational | Basic information about the project goals and current status | Y |
| Training | Dissemination of tutorials, educational materials, annotation guidelines and SOPs | Na |
| Download | Repository for project sequence and annotation data | Y |
| Tools | ||
| Genome browsers | Analysis of genomic context | Y |
| SkateBLAST | Searching and download of genomic contigs and features | Y |
| SkateBase community | Connectivity and coordination of community annotation activities | Na |
| File exchange | Sharing of raw and analyzed high-throughput sequence data | N |
| RACE-P | Community annotation of proteins | Y |
aFeature under development for future public release.
Little Skate Genome Annotation workshops
A model of collaborative and distributed training was employed for skate genome annotation. Three face-to-face genome annotation workshops were organized, with the aim of developing a knowledgeable workforce in the cutting-edge genomic and bioinformatic sciences, and to foster data-intensive collaborative research across the region (http://bioinformatics.udel.edu/research/skategenomeproject). A timeline of the annotation workshops and genome-sequencing progress is shown in Figure 2. These 1-week-long workshops were held in Delaware and Maine and were attended by participants from all five NECC states. Each workshop was built upon its predecessors, while containing standalone training modules to prepare new workshop participants for hands-on bioinformatics activities. The first workshop, hosted by UD in May 2010, covered all aspects of genome sequence analysis required to annotate eukaryotic genomes. The second and third workshops, held at the MDIBL in October 2010 and UD in May 2011, respectively, focused on hands-on annotations of the emerging skate sequence data. The third workshop was held in conjunction with UD's Research Symposium on Bioinformatics and Systems Biology, providing opportunities for further scientific exchange among participants. The instructors included NEBC bioinformatics scientists and other invited experts in the field.
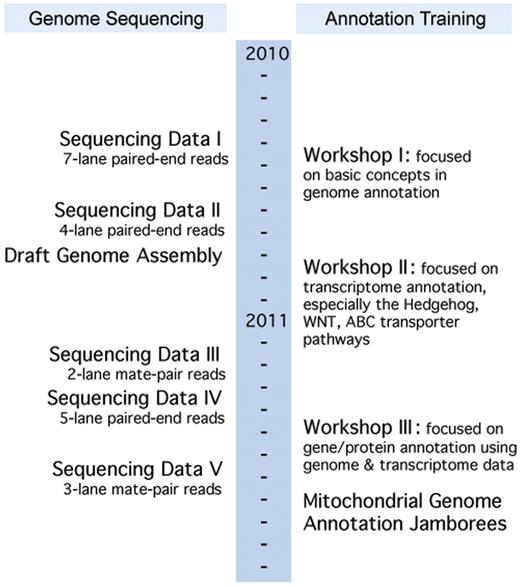
Little Skate Genome Project's timeline indicating the simultaneous annotation training and genome development. Sequencing Data Sets I: seven lanes of paired-end reads; II: four lanes of paired-end reads; III: two lanes of mate-pair reads; IV: five lanes of paired-end reads; V: three lanes of mate-pair reads. There are a total of 2 931 925 134 reads.
In preparation for the workshop, pre-computed BLAST (blastx) searches of the L. erinacea genomic contigs and the transcriptomic data against a database of all vertebrata protein sequences in UniProtKB (18) were conducted to produce an initial gene set for the participants to choose for the annotation exercises. During the hands-on activities, participants started with one candidate gene and conducted reciprocal BLAST searches to identify homologous proteins between L. erinacea and other vertebrates. Based on this, participants provided gene annotations that included: (i) gene name, (ii) evidence of a single transcript with complete coding sequence, (iii) exons identified in the genome contig and (iv) gene structure compared with that of human and mouse. The wiki-based SkateBase Community tool was used to assist novice users in adding and editing annotations. The annotation results can be modified by the participants and instructors, and become visible and searchable to instructors immediately, allowing timely review and feedback. In addition to the gene annotation, participants annotated L. erinacea protein sequences available in UniProtKB (18) using the Race-P annotation interface.
Between the three workshops, 56 trainees received instruction and hands-on experience annotating genomic sequence data. Training was led by 10 instructors from NEBC institutions, as well as 14 guest lecturers from government, academia and industry, including NCBI, Protein Information Resource (PIR), Illumina and the University of Virginia. On average 32 h of training were provided by each workshop, with several participants receiving nearly 100 h of experience by attending all three workshops.
Mitochondrial Genome Annotation Jamborees
The Mitochondrial Genome Annotation Jamborees aimed to annotate all the genes and regulatory regions in the completed mitochondrial genome of L. erinacea, providing a culminating experience following the three annotation workshops and serving as a model for our continuing collaborative annotation activities. The 29 participants ranging from classes of undergraduates to junior faculty were led in these activities by seven different instructors, two of whom participated in past workshops as trainees.
This community annotation process started with an introductory presentation webcasted from UD to participating institutions across the NECC states, giving all participants a common background on the project, its goals, vertebrate mitochondrial genomes and the annotation tools. While adopting the same overall workflow and bioinformatics framework (Figure 3), each state customized their workflow to accommodate specific needs.
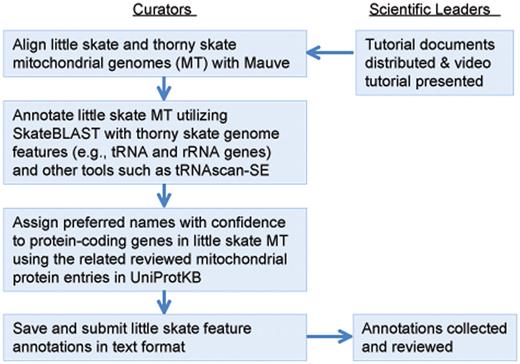
Mitochondrial genome annotation jamboree workflow. Curators from each state worked independently for ∼2 weeks before submitting results to project leaders for review.
Following the introductory session, participating curators from each state were given 1–2 weeks to finish their individual annotation of the 39 gene features, with tutoring available during the period. This is followed by discussions among curators and the scientific leaders within their state to reach a group consensus for annotation. Finally, the scientific leaders from all participating states collated and discussed the annotation results from various states through video conferences. Discrepancies were discussed and other data sources such as tRNAscan-SE (19, 20) were considered to finalize the annotations and provided feedback to participants. Feedbacks were provided to participating annotators.
The SkateBase web portal provided a central access to various analysis and annotation tools, along with specific resources needed for the mitochondrial genome annotation. The latter include: (i) the sequence and annotation files for little skate and its relatives—the thorny skate (Amblyoraja radiata) (21), and ocellate spot skate (Okamejei kenojei; also known as Raja porosa) (22); and (ii) databases containing mitochondrial features from A. radiata and O. kenojei and the 13 mitochondria protein families derived from UniProtKB for homology searches using SkateBLAST.
This community genome annotation has been introduced into an educational curriculum, as demonstrated by a cross-disciplinary course at the University of Rhode Island (URI), ‘Practical Tools for Molecular Sequence Analysis’, designed to teach bioinformatics to students in the biological sciences. Following the initial lecture and annotation sessions, variability in gene annotation was corrected in subsequent sessions. Students gained experience in constructing a custom BLAST database from A. radiata and O. kenojei mitochondrial genome features to query against L. erinacea mitochondrial genome. Additionally, participants conducted a comparative analysis using the Geneious software package (Biomatters Ltd., Auckland, New Zealand) to identify discrepancies in the gene structure.
Little skate mitochondrial genome annotation results
The consensus annotation of genes and other features from this community annotation for the L. erinacea mitochondrial genome are similar to typical vertebrate mitochondrial genomes including other elasmobranch fishes (23). There are 13 protein-coding genes, 22 tRNA and two rRNA genes along with two miscellaneous sequence features in the 16 724-bp genome (Figure 4A). The order and orientation of the features along the chromosome are the same as the mitochondrial genomes of two other skate species, A. radiata (21) and O. kenojei (22) (Figure 4B). In general, the L. erinacea genome is more similar to A. radiata than to O. kenojei in terms of sequence similarity for genes and other features. However, the overall length of the L. erinacea mitochondrial genome is closer to O. kenojei as the intergenic region between tRNA-Thr and tRNA-Pro is longer in A. radiata than the other two species.
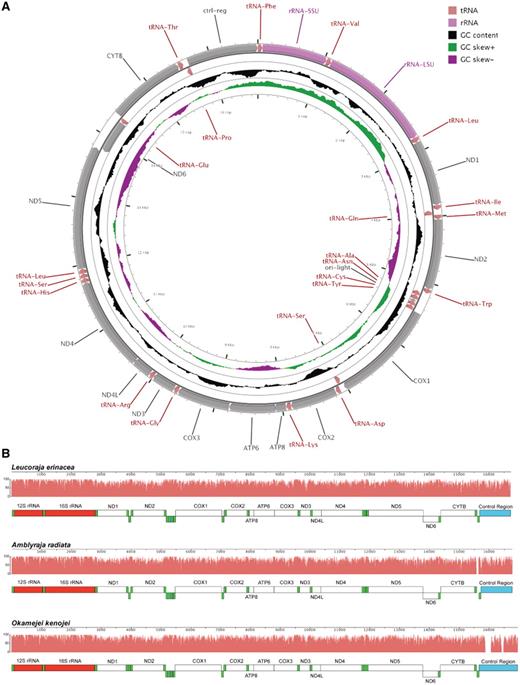
Leucoraja erinacea mitochondrial genome. (A) Leucoraja erinacea mitochondrial genome with the consensus annotation for genes and other sequence features generated using CGView (29). The orientation of genes is shown with arrow heads. The tRNA genes are shown in pink, rRNA genes in purple and protein-coding genes in grey. The first inner circle shows the GC content above and below the average GC content for the mitochondrion in black. Positive GC skew is shown in green and negative in magenta. (B) The mitochondrial genomes of L. erinacea, A. radiata and O. kenojei are displayed using Mauve (16, 17), with rRNA features in red, tRNA features in green, protein-coding regions in white, and miscellaneous features in blue. The pink profiles indicate the sequence identity levels among the three genomes.
Similar to other vertebrate genomes, there are 12 protein-coding genes encoded on the heavy strand and one (ND6) on the light strand. These genes are highly conserved, with encoded proteins of >95% identity to orthologs in A. radiata. In a few cases (ND1, ND2, ND5 and COX2), relying on tBLASTn alignments was insufficient since coding sequences could not be properly determined without an additional analysis of ORF.
The 22 tRNA genes in L. erinacea are highly conserved with other vertebrate genomes in terms of their order and orientation. There are two tRNAs for leucine and serine like other vertebrate genomes. The intergenic region between tRNA-Thr and tRNA-Pro was annotated in A. radiata to be 68 bp and contain a putative tRNA (21). In L. erinacea, this region is just 9 bp which is similar to the 6-bp length observed in O. kenojei. All tRNAs were identified using both sequence similarity and gene prediction via tRNAscan-SE (19, 20) except for the tRNA-Ser between tRNA-His and tRNA-Leu that was not predicted. This tRNA was 91% (62/68) identical to A. radiata and 83% (57/69) to O. kenojei with one gap.
The 12S and 16S rRNA genes are highly conserved. Alignment of 12S from L. erinacea to the orthologs in A. radiata and O. kenojei showed 98 and 95% with three gaps, respectively. For 16S, the identity was 98% with one gap and 93% with eight gaps, respectively.
The two miscellaneous sequence features annotated were the control region and the light chain origin of replication. The control region in L. erinacea aligned to the region from A. radiata in one large block of 1058 bp each at 91% identity. Alignment to the region from O. kenojei showed two large blocks of 473 and 486 bp each with 85 and 75% identity, respectively. The light chain origin of replication was 100% identical to both A. radiata and O. kenojei as expected.
Dissemination
To date, the Little Skate Genome Project has resulted in a draft assembly of 11 lanes of Illumina paired-end sequencing data producing 2 962 365 contigs (N50 = 665 bp) totaling 1.5 Gb of DNA sequence (almost one half of the expected genome size), with the longest contig 21 kb. An additional 10 lanes of Illumina HiSeq mate-pair and paired-end sequencing has been completed and awaits inclusion in the upcoming draft assembly build 2. In addition to the assembly and annotation of the 39 features of the 16.5 kb mitochondrial genome reported here (GenBank Accession: JQ034406), these data have already contributed to two high-impact publications (12, 24).
To further expand on this successful utilization of the project's data, dissemination has been identified as a top priority. As noted elsewhere, raw sequence data, the draft genome assembly build 1, and mitochondrial genome annotations are made available as part of genome project PRJNA60893. The SkateBase website developed by this project, further acts as a centralized repository for these sequence and annotation data, additional project results and metadata, tools for visualizing and searching these data, project news and educational content (Table 1). Dissemination is an ongoing process that will continue to evolve with the increasing output of the Little Skate Genome Project.
Summary and future directions
Lessons from the Little Skate Genome Annotation Workshops and Jamborees
The collaborative processes devised and lessons learned from the annotation workshops and jamborees discussed herein have served as an infrastructure-building model that will be used extensively for the continuing annotation work on the little skate genome. Feedback from participants in all three workshops was very positive overall. The feedback from earlier workshops helped improve later workshop to provide a better learning experience for participants, such as shorter lectures on background material coupled with more extensive hands-on activity. Indeed coupling training with annotation has fostered better understanding of the tools taught in lectures. The hands-on exercises with real-world problems provoked deeper thinking and strengthened understanding of abstract bioinformatics concepts.
One challenge in organizing the genome-annotation workshops was the disparate background of the participants, which included faculty, postdoctoral fellows and graduate and undergraduate students from diverse fields. Consistent with the goal of the workshops for workforce development, prior bioinformatics knowledge or experience was not a prerequisite to attend the workshops. The curriculum was thus designed to allow novice researchers to learn about bioinformatics and genome annotation, while hands-on activities allowed more experienced participants to explore in-depth annotation. Annotation activities paring experts and non-experts (or senior and junior participants) in small groups have facilitated active learning via peer mentoring. Also critical to the success of the annotation workshops and jamborees for such diverse participants are training materials including tutorials and clear annotation guidelines, as well as intuitive web-based annotation interface with analysis and visualization tools.
A second challenge was recruiting participants and motivating attendance to the workshops. While a recent survey (25) suggested that ‘reward’ and ‘employer/funding agency recognition’ were less important than awareness of the community annotation opportunity and ease-of-use of annotation interfaces, a follow-up discussion nevertheless pointed out the importance of incentives [see Reviewers’ comments and Authors’ response in ref. (25)]. To promote broad community participation, the NEBC workshop committee did provide a number of incentives, including full funding to attend the workshops including travel and lodging, free advanced training in bioinformatics, visit to state-of-art sequencing facilities, poster presentations, meeting and interacting with multidisciplinary researchers with a common interest in bioinformatics, and a certificate at the end of workshops. Moreover, biocuration as an emerging and fast-growing career was introduced to the broad audience (26). Authors of annotated entries received proper credit for their contributions on the SkateBase website, and will be acknowledged through the public dissemination of those entries.
A third challenge was the active participation by students, especially undergraduates during regular semesters due to their course load. Accordingly, two annotation workshops were held just after the end of classes in spring semesters to accommodate their schedules. Another alternative is to engage students through independent research or special topics courses. The direct integration of genome annotation into regular biological sciences courses as has been shown at URI also provides effective means for student training and participation.
Three sources of conflicts were observed upon review of the annotated gene and sequence feature coordinates. First, many participants only used BLAST sequence similarity searching to annotate the protein-coding genes instead of also looking for ORFs. As described above, the sequence similarity for ND3 gene in the three skates was low near the end of the sequences (Figure 4B), resulting in an alignment that did not extend over the entire length of the gene. Second, inconsistency between annotations of tRNA coordinates in the A. radiata and O. kenojei genomes was one source of conflicting annotations among participants. Since the O. kenojei mitochondrial genome has been annotated by the Reference Sequence Project at the NCBI, these conflicts could be readily resolved by examining a tRNA gene in this genome. Lastly, simple typographical errors of keying in the coordinates were observed in some annotations.
A number of important lessons were learned for undergraduate and graduate participation during the classroom sessions. Common cognitive obstacles were navigating among the multiple web-based resources, forgetting about the unique codons in the mitochondrion and keeping track of the position numbers of the sequences being compared. Many students were unable to construct their own workflow of the needed resources and instructor demonstrations of the process were generally insufficient. Thus, an introductory presentation and providing guided structure as they worked their problems was essential. In addition, the students were reluctant to double-check their results, and, unsurprisingly, were uncomfortable with ambiguity. In the RI graduate class, a second session in which the students repeated the annotation exercise but using a different environment (command-line BLAST instead of web-based tools) reinforced the basic principles of sequence annotation and allowed for corrections of many simple errors.
For community intelligence applications to achieve the critical mass of users and activity, a positive feedback loop with three components is needed, according to previous findings (27): scientific utility, community usage and community contributions. There is already interest in this annotation effort shown from a couple of research groups in institutions outside of NECC states such as Stanford University and the University of Maryland. In terms of community usage, the workshop lectures and other materials (e.g. SkateBase tools) will serve as a valuable resource for a broad user base on which the future skate Genome community annotation initiative will build. Within NECC states, the training materials are being integrated into the educational curricula across institutions, such as a class taught by workshop participant Shallee Page at the University of Maine at Machias (class: Introduction to biochemistry class), and workshop participant and state-coordinator Dan Udwary at the University of Rhode Island (class: Practical Tools for Molecular Sequence Analysis).
Future directions
The importance of continuing to improve the assembly cannot be overstated as the quality of annotations and sequence analyses are directly proportional to the quality of the underlying sequence. The annotation efforts will continue, with support, accessibility and content improvement of current tools.
Overall, providing a user-friendly community annotation platform, with easy-to-use interfaces plus simple and clear instructions on what to annotate as suggested in (25), will be central tasks for Little Skate Genome Project to harness the principle of community intelligence, enabling any user to easily and directly contribute to the annotation. Powerful user customizability may be another factor to consider when implementing a user interface. The activities to date serve as learning exercises for annotators and organizers, as well as a test of infrastructure built to promote this project. Lessons and skills learned through these early exercises will enable more productive community annotation effort going forward on this project. For example the activities have suggested that incorporation of the annotation assignments into educational curricula across institutions can be an effective means for obtaining quality annotations through semester-long or academic year-long training and assessments.
Materials and methods
Sample collection
A genomic DNA sample from a single L. erinacea Stage 32 embryo (Marine Biological Laboratory, Woods Hole, MA, USA) was prepared by Dr Carolyn Mattingly at MDIBL. Tissue was frozen and ground in liquid nitrogen and genomic DNA was extracted using the Gentra Puregene kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol.
Sequencing
The DNA sample was sent to the Sequencing and Genotyping Facility at the University of Delaware for DNA library preparation and Illumina-based sequencing. For paired-end library preparation, genomic DNA was fragmented to a uniform size of ∼500 bp using the Covaris S2 Acoustic Disruptor (Covaris Inc., Woburn, MA, USA). For mate-pair library preparation, genomic DNA was fragmented to generate uniform fragment sizes of 2.5 kbp, 3.5 kbp and 5 kbp using the Hydroshear (Digilab Inc., Holliston, MA, USA). Sequencing libraries were prepared using conventional Illumina paired-end and mate-pair library preparation kits (Illumina Inc., San Diego, CA, USA). Five hundred basepair paired-end libraries were clustered on the Illumina Cluster Station and subsequently sequenced on the Illumina GAIIx platform. Mate-pair libraries were clustered on the Illumina cBot and sequenced on the Illumina HiSeq2000 platform. Sequencing protocol used for paired-end libraries was 2 × 125 cycles. Sequencing protocol used for mate-pair libraries was 2 × 125 cycles for the 3.5-kb library, and 2 × 50 cycles for the 2.5- and 5-kb libraries. Cluster identification, base calling and quality scoring were performed using Illumina Sequencing Control Software and Real Time Analysis. FastQ files were generated from base calls using the Illumina CASAVA pipeline. A total of 2 534 435 707 sequence reads were generated using 16 Illumina flow-cell lanes completed as of October 2011.
Assembly
The mitochondrial genome contig was assembled using CLC Bio Genomics Workbench 4.6 (CLC Bio, Aarhus, Denmark). A subset of reads consisting of all paired-end reads and the 3.5-kb mate-pair library were assembled using default settings of the Genomics Workbench to produce 3 million contigs. One hundred and one of these contigs were over 10 000 bp in length. The mitochondrial genome was found in this set of long contigs.
Data deposition
Little Skate Genome Project sequence and annotations are collected under GenBank BioProject 60893. Contig sequences (Draft Assembly Build1) utilized in the workshops and jamborees reported here are available from GenBank (AESE010000000) and SkateBase (http://skatebase.org/downloads). The complete raw data has also been submitted to NCBI's Sequence Read Archive (SRA026856). The mitochondrion genome sequence and annotation is available through GenBank Accession JQ034406. Additional annotation, metadata and other project information is made available through SkateBase.
Annotation
Gene annotation was performed as detailed in ‘Mitochondrial Genome Annotation Jamborees’ section. In addition to the SkateBase sharing and annotation tools described in ‘Collaborative Tools’ section, several other software tools were utilized to provide additional annotation evidence and advanced visualization. The program tRNAscan-SE was used with default settings for organelle mode to confirm boundaries and locations of tRNAs (19, 20). The Mauve progressive multiple genome aligner was used to provide comparative alignment of genome features from L. erinacea, A. radiata and O. kenojei (16, 17). The multiple sequence alignment server in PIR was also used (28).
Funding
This work was supported by linked grants from the National Center for Research Resources, National Institutes of Health (P20RR16462 for Vermont Genetics Network - Vermont INBRE (IDeA Networks of Biomedical Research Excellence), P20RR016463 for Comparative Functional Genomics INBRE in Maine, P20RR016457 for Rhode Island INBRE, P20RR018787 for Cellular and Molecular Mechanisms of Lung Disease, P20RR016472 for Delaware INBRE), as well as the Experimental Program to Stimulate Competitive Research (EPSCoR), National Science Foundation (EPS-0918284 for University of Vermont, EPS-0918033 for University of New Hampshire, EPS-0918078 for University of Delaware, EPS-0918018 for University of Maine, and EPS-0918061 for University of Rhode Island). The participant costs of the first and third annotation workshops were funded by the 3P20RR016472-09S2 Delaware INBRE Administrative Supplement. Funding for open access charge: NIH (P20RR016472 for Delaware INBRE).
Conflict of interest. None declared.
Acknowledgments
We are grateful to the speakers for the Little Skate Genome Annotation Workshops: Drs David Landsman, Deanna Church and Kim Pruitt, at National Center for Biotechnology Information, National Institutes of Health; Dr Jason Moore at Dartmouth Medical School; Dr William Pearson at University of Virginia; Drs. Randall D. Dahn, Tony Planchart, Jim Coffman and Carolyn Mattingly at MDIBL; Dr Carol Bult at Jackson Laboratory; Mr Craig Fishman at Illumina; Drs Karl Steiner and Mihailo Kaplarevic at University of Delaware; Dr Joanna Fueyo at University of Rhode Island; Drs Raja Mazumder and Sona Vasudevan at Protein Information Resource. Drs Karol Miaskiewicz and Mihailo Kaplarevic are acknowledged for their support for the NECC Shared Data Center housed at UD. We thank Dr Carolyn Mattingly for preparing the skate genomic DNA sample and Dr Randall D. Dahn for skate transcriptome data. We also thank Ms Susan Phipps and Katie Lakofsky for their assistance during the two workshops at UD.
The North East Bioinformatics Collaborative (NEBC) Curation Team includes DE: Daniel Nasko, Chandran Sabanayagam, Liang Sun and Yue Wang at University of Delaware; ME: Jacob Berninger, Stevey Mahar, Eric Tan and John J. Wilson at University of Maine at Machias; Vanessa Coats at University of Maine; Clare Bates Congdon, Jeffrey Ahearn Thompson and David J. Gagne at University of Southern Maine; RI: Jimmy Adediran, Thomas Bregnard, Alison C Cleary, Scott Grandpre, Bethany Jenkins, Lauren Killea, Bradford Lefoley, Katherine Mccusker, Matthew Mokszycki, Megan O'Brien, J.Christopher Octeau, Steven Shelales, Edward Spinard, Jacob Stupalski, Linh Tran, Joselynn Wallace at University of Rhode Island; VT: Brian Cunniff at University of Vermont.
References
Author notes
†These authors contributed equally to this work.
‡The members of the North East Bioinformatics Collaborative (NEBC) Curation Team are provided in the ‘Acknowledgements’ section.

{kind=link}
{kind=link}
{kind=link}
{kind=link}