





Abstract
The overwhelming fraction of proteins whose sequences have been collected in comprehensive databases may never be assessed for function experimentally. Commonly, putative function is assigned based on similarity to experimentally characterized homologs, either on the level of the entire protein or for single evolutionarily conserved domains. The annotation of individual sites provides more detailed insights regarding the correspondence between sequence and function, as well as context for the interpretation of sequence variation and the outcomes of experiments. In general, site annotation has to be extracted from the published literature, and can often be transferred to closely related sequence neighbors. The National Center for Biotechnology Information's Conserved Domain Database (CDD) provides a system for curators to record functional (such as active sites or binding sites for cofactors) or characteristic sites (such as signature motifs), which are conserved across domain families, and for the transfer of that annotation to protein database sequences via high-confidence domain matches. Recently, CDD curators have begun to sort-site annotations into seven categories (active, polypeptide binding, nucleic acid binding, ion binding, chemical binding, post-translational modification and other) and here we present a first comparative analysis of sites obtained via domain model matches, juxtaposed with existing site annotation encountered in high-quality data sets. Site annotation derived from domain annotation has the potential to cover large fractions of protein sequences, and we observe that CDD-based site annotation complements existing site annotation in many cases, which may, in part, originate from CDD's curation practice of collecting sites conserved across diverse taxa and supported by evidence from multiple 3D structures.
Introduction
The Conserved Domain Database (CDD) (1) is a manually curated protein annotation resource developed and maintained by the National Center for Biotechnology Information (NCBI). CDD collects a large set of protein and protein domain models, as multiple sequence alignments and derived position-specific score matrices (PSSMs), and uses RPS-BLAST (2), a variant of the widely used PSI-BLAST algorithm (3), to match protein database sequences with these family models. While the majority of models are imported from external sources, the CDD curation team is revisiting larger protein domain superfamilies to establish finer-grained hierarchical classifications that are based on phylogenetic analysis and supported by the published literature, functional annotation, domain architecture and taxonomic distribution. While characterizing individual subfamilies, curators also record conserved functional sites and evidence for those sites, in a way so that sites can be mapped onto protein sequences using pre-computed protein-model alignments as collected in the Conserved Domain Architecture Retrieval Tool (CDART) database (4). CDD-based site annotation is readily visible on Entrez's GenPept summary pages for proteins and in graphical views (Figure 1), and it is being distributed via NCBI's Reference Sequence protein data sets (5). More recently, CDD site annotation is used to verify and rank clusters of interactions observed in 3D structures as presented by the Inferred Biomolecular Interactions Server (IBIS) resource (6), where such clusters can be used to infer interactions for proteins sequence similar to those with known 3D structure. CDD site annotation is also visible in the domain mapping of disease mutations (DMDMs) resource, where it can be contrasted with known disease mutations and polymorphisms (7).
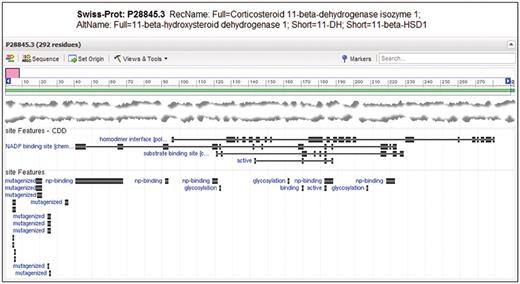
Entrez Protein graphical sequence view for SwissProt sequence P28845.3, gi|118569. At the bottom of the view, site annotation (labeled ‘site Features’) from CDD and as encountered in the original record are visible on top of each other. Note that CDD annotates the homodimerization interface, substrate and cofactor binding sites and active site as relatively large sets of disjoint residue positions. The homodimer interface annotation is not present in the original annotation, but it provides unique labeling of glycosylation sites.
SwissProt, as maintained by the UniProt Knowledgebase (8), is a resource that provides high-quality manually curated annotation of protein sequences. SwissProt-annotated sequences are tracked by NCBI's Entrez protein database, including the site annotation provided by the source data. Here, we present a study that examines a subset of the SwissProt-based sequences tracked by Entrez, namely those already covered by NCBI-curated domain models, and compares site annotations that originate from CDD with annotation originating from SwissProt.
Conserved domain site annotation
The curation of domain models in CDD aims at characterizing protein domain superfamilies as collections of sequence fragments related by common evolutionary descent, organized into multiple sequence alignments and split into subfamilies that reflect ancient gene duplication events and subsequent divergent evolution. Curation of CDD-conserved domain hierarchies has been explained in previous manuscripts (9). Typically, a domain subfamily is created and annotated if it is supported by phylogenetic analysis and contains member sequences from diverse organisms, suggesting an origin several hundred million years in the past. To this end, curators compute and examine sequence tree displays, to select robust branches and will consider taxonomic distribution, domain architecture, protein annotation and existing/external classifications. CDD curators make extensive use of protein 3D structure, when available, as in-house curation tools are tightly coupled to the Entrez 3D structure database Molecular Modeling Database (MMDB) (10) and structure neighboring data computed with Vector Alignment Search Tool (VAST) (11), and the associated 3D viewer Cn3D (12) is the main alignment viewing and editing tool. From examining patterns of sequence conservation, the published literature, and the 3D structures of complexes that may contain proteins interacting with binding partners, curators often notice and record the location of functional sites or motifs characteristic for a domain family. Sites are recorded as addresses on the multiple sequence alignment models that describe the domain family, and this mapping is being transferred into the coordinates of the PSSMs that are used to scan the protein sequence database. From an alignment of a protein sequence to a PSSM, the site coordinates can be again transferred onto the protein sequence itself. This is only done if the mapping of the site is near complete; partially aligned sites are not used to infer sites on protein sequences. Functional sites associated with a domain model are only mapped onto proteins with high-scoring-specific hits to that model. Sites are recorded with a short name, such as ‘active site’ or ‘ATP binding site’. Although common site names are now being selected from a list of pre-defined expressions, the name is stored as free text and can be modified by the curators as they see fit. We have recently started to assign site types and to retrofit existing models with site-type definitions. CDD deliberately picked a small number of seven generic site types, so that the majority of annotations that we will come across can be sorted into the seven types in a straightforward manner. The site types were also selected to match the IBIS classification of interaction sites (6), as CDD curators use IBIS in the curation work flow. Curators pick common site names from a small set of pre-defined and generic options (such as ‘active site’ or ‘dimerization interface’), but also refer to the literature when deciding on a site name, and are free to choose very specific names if deemed appropriate. The site types used in CDD are listed in Table 1.
Site types and names as defined in Conserved Domain Database models and as mapped onto protein sequences in Entrez
| Type designation | Examples of common names | Counts |
|---|---|---|
| Active | Active site, catalytic site | 3300 |
| Polypeptide binding | Dimer interface, oligomer interface | 3020 |
| Nucleic acid binding | DNA binding site, RNA binding site | 482 |
| Ion binding | Ca binding site, Zn binding site | 1500 |
| Chemical binding | ATP binding site, NAD(P) binding site | 3310 |
| PTM | Glycosylation site, phosphorylation site | 104 |
| Other | Walker A/P-loop, activation loop | 4439a |
The counts reflect the numbers of site annotations recorded on CDD models in the most recent release, v2.32.
aNote that sites without any explicit alternative type assignment are flagged ‘other’; as site typing is an ongoing process, this number reflects models that still need to be revisited more than the actual fraction of sites that cannot be sorted into a more specific category.
Curators also record evidence together with the conserved site annotation, which is presented to CDD users via conserved domain summary pages. Evidence may be free text comments, references to journal articles or structure evidence, which contains instructions for highlighting a site in a particular 3D structure used in the model, together with a binding partner that exemplifies the biological significance of the site annotation.
Conserved sites are annotated only if it seems reasonable to assume that the site is present in all or nearly all sequence fragments specifically annotated by the respective model. Mapping of sites via homologous relationship will undoubtedly generate false annotation, but that fraction is expected to be small if (1) site annotation is restricted to well-conserved motifs that are linked to the generic function of the domain family, and (2) a conservative procedure is used to qualify a match for mapping sites. Consequently, site annotation in CDD is restricted to sites that tend to be well conserved in divergent evolution. It is evident from Table 1 that relatively few post-translational modification (PTM) sites have been recorded, for example, as these tend to evolve rather quickly and are often not associated with the structurally conserved core segments of conserved domains, which constitute the bulk of CDD's alignment models. The low number of PTM sites is most likely due to the lack of conservation between sites in a single domain model; their annotation would require further fine-grained subfamily classification, as curators only annotate sites that appear conserved in all or nearly all representative sequences of a domain model.
Specific domain hits and site mapping
The collection of domain models in CDD is redundant, as CDD mirrors several external resources. It is quite common to have the same domain family described by models from three or four different sources, and if hierarchical classifications of diverse superfamilies are available, dozens of models may provide overlapping annotation for a particular region on a protein. To deal with this redundancy, CDD presents a simplified default view of domain search results: models describing homologous families are grouped together into superfamily clusters, and the annotation with a superfamily cluster is presented instead of the single model that happened to score the best hit. However, if the highest ranked hit was scored by an NCBI-curated model, and that score exceeds a model-specific threshold, (13) the ‘specific hit’ is presented on top of the superfamily annotation. CDD follows simple rules for mapping site annotations onto protein sequences: functional sites associated with a domain model are only mapped onto proteins with high scoring-specific hits to that model. If only a superfamily annotation is shown, but if the set of redundant hits includes an NCBI-curated model, site annotation is mapped from the root node of the conserved domain hierarchy that model came from—annotating only the most generic sites that are presumed conserved across the entire superfamily.
Methods
A subset of NCBI's Entrez protein sequence records contain site annotation provided by the originating source database. For the analysis presented here, we chose to use sequences that are flagged as originating from SwissProt. Sixty-six percent of all SwissProt sequences in Entrez/protein had site annotation from some source; two-thirds of these had hits to specific CDD-curated domain models; ∼45% of all SwissProt records had such specific hits. We focused the analysis on the latter, SwissProt sequences that had specific domain annotation from CDD, meaning that at least one sequence region comes with high-confidence identification of a conserved domain, which may also include mapped site annotation. This restricts the analysis to a set of protein domain families that have undergone curation by CDD staff to date, and it results in 233,722 sequences (as of September 2011). Site annotations in those sequences were collected, including the site type assigned in each case. Pre-existing (non-CDD) site annotation, which was interpreted as stemming from the SwissProt curation effort, is categorized into a larger set of 12 site types in Entrez, which reflects the site typing undertaken by curatorial staff at the source database, while CDD-based site annotation uses the 7 types outlined in Table 1. We defined two sites from different sources as overlapping if they shared one or more residue coordinate on the protein sequence. In the analysis presented below, we did not try to map site types between CDD and Entrez/protein.
Results and conclusions
CDD maps site annotation onto several million proteins in Entrez. Figure 2 presents the site annotation coverage for the subset analyzed in this manuscript.
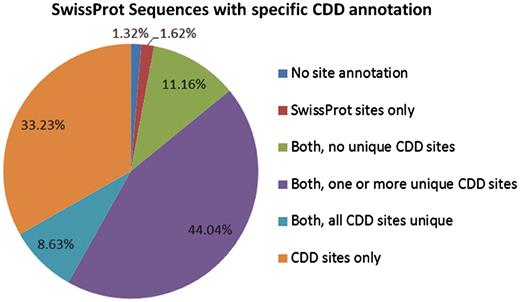
The 233 722 protein sequences we analyzed can be categorized based on the source of site annotation. A small number, 1.32% of the SwissProt sequences with specific hits to NCBI-curated domain models, do not have any site annotation. The 1.62% have site annotation only from SwissProt, and 11.16% have CDD site annotation that appears redundant (overlaps with existing SwissProt annotation). For the remaining 85.9%, CDD provides some unique site annotation, and for about one-third of the sequences CDD provides the only site annotation.
It seems evident that CDD site annotations contribute to a large fraction of the proteins that are covered by the current curation effort. Of the 1 491 437 individual site annotations we tracked, just a little more than half (53.3%) came from mapping of CDD sites, and they are spread across 97% of the sequences in the set, reflecting the fact that the majority of NCBI-curated domain models do also come with functional site annotation. In more than half of the proteins, some or all of the CDD annotation overlaps with annotation provided by SwissProt, but CDD also contributes unique sites, and sometimes the only site annotation available at this point. Figures 3 and 4 detail the distributions of site annotations according to the assigned site type, for CDD and SwissProt, accordingly.
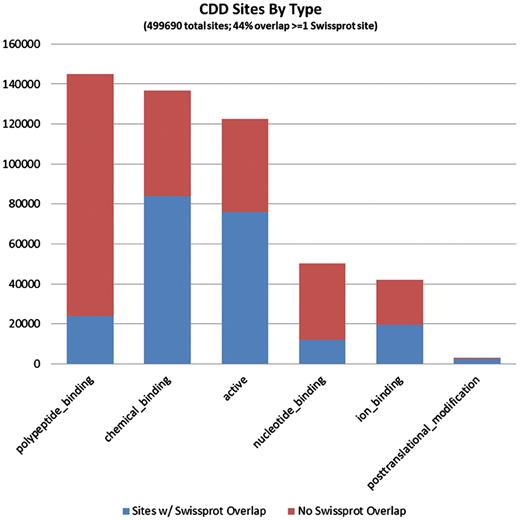
The 794 228 site annotations on protein sequences we analyzed, which were generated via mapping to CDD models, can be categorized based on the site type assigned by CDD. A large fraction of sites is assigned type ‘0’ or ‘other’, as the typing of all previously recorded sites has not been completed. These are not shown here. CDD annotates only a small number of PTM sites, as these are rarely conserved across somewhat diverse domain families. The bars are colored according to the overlap with SwissProt sites (irrespective of the SwissProt site type). It appears that polypeptide-binding sites, those conferring protein–protein interactions, are most often uniquely annotated by CDD.
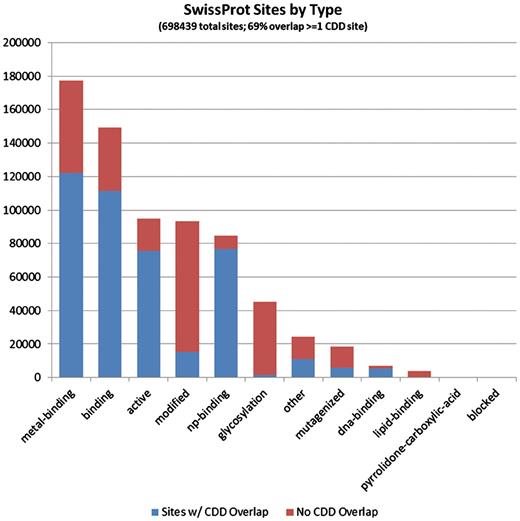
The 697 209 site annotations encountered on the protein sequences we analyzed, which originate from the SwissProt curation effort, categorized based on the site type assigned in Entrez/protein. The bars are colored according to the overlap with CDD-generated sites (irrespective of the CDD site type). It appears that PTM sites, those summarized under the ‘modified’ and ‘glycosylation’ types, are most often uniquely annotated by SwissProt.
While there is a large degree of overlap between CDD-generated site annotation and SwissProt-generated annotation, we notice that the two data sources also complement each other to a certain degree; for ∼33% of the SwissProt sequences with specific CDD domain annotation, CDD provides the only site annotation. Individual sequence curation—and inference of sites between close homologs—can record the presence of functional sites that are not conserved across more diverse families. The comparative analysis of protein 3D structure complexes, on the other hand, enables CDD curators to record the positions of interfaces with which macromolecules interact, including homo and hetero-oligomerization interfaces. It may be helpful to consider both sources of annotation in the study of protein function and the design of experiments, so as to benefit from curation work approaching the issue from different angles.
The strength of CDD's approach is that conserved sites can be annotated on large numbers of protein sequences with relatively little effort, as a single model may provide ‘specific domain hits’ to hundreds or thousands of protein sequences. Naturally, this will also lead to a higher incidence of false positive annotation. We are in the process of implementing curation software that allows for conditional functional sites: curators will be able to specify the amino acid residue types that are allowed in selected positions of a functional site. Consequently, sites will be only mapped onto sequences if the site address matches such a defined sequence motif that is associated with known or proven function. While this is expected to reduce the incidences of false annotation, it will be particularly useful for annotating sites that are known as not strictly conserved across all sequences that define a domain family, such as PTM sites.
Feedback with respect to inaccurate site annotation or supporting and conflicting experimental evidence is welcome and concerns can be addressed efficiently via the CDD curation pipeline.
Funding
This work was funded by the Intramural Research Program of the National Library of Medicine at the National Institutes of Health/DHHS. Funding for open access charge: Intramural Research Program of the National Library of Medicine at National Institutes of Health/DHHS. Comments, suggestions, and questions are welcome and should be directed to: info@ncbi.nlm.nih.gov.
Conflict of interest. None declared.
Acknowledgements
We thank the Conserved Domain Curators for compiling the site annotations analyzed in this work, Farideh Chitsaz, Noreen Gonzales, Marc Gwadz, Fu Lu, Gabriele Marchler, James Song, Narmada Thanki, Roxanne Yamashita, Chanjuan Zheng, as well as the CDD alumni Anastasia Nikolskaya, Raja Mazumder, Natalie Fedorova, Aviva Jacobs, B. Sridhar Rao, Sona Vasudevan, Luning Hao, Jodie Yin, Dmitri Krylov, Asba Tasneem, Zhaoxi Ke, Mikhail Mullokandov, Marina Omelchenko, John Jackson, John Anderson, Cynthia Robertson and Carol DeWeese-Scott. We thank Renata C. Geer for assistance with preparing figures.

{kind=link}
{kind=link}
{kind=link}
{kind=link}