





Abstract
Current high-throughput technologies for investigating genomic variation in large population based samples produce data on a scale of millions of variations. Browsing through these results and identifying relevant functional variations is a major hurdle in these genome-wide association studies. In order to help researchers locate the most promising associations, we have developed a web-based database portal called Varietas. Varietas can be used for retrieving information concerning genomic variations such as single-nucleotide polymorphisms (SNPs), copy number variants and insertions/deletions, while enabling users to annotate large number of variations in a batch like manner and to find information about related genes, phenotypes and diseases. Varietas also links out to various external genomic databases, allowing users to quickly browse through a set of variations and follow the most promising leads. Varietas periodically integrates data from the major SNP and genome databases, including Ensembl genome database, NCBI dbSNP database, The Genomic Association Database and SNPedia.
Database URL:http://kokki.uku.fi/bioinformatics/varietas/
Introduction
The growth in popularity of high-throughput technologies for identifying genomic variations such as single-nucleotide polymorphisms (SNPs), insertions/deletions and copy number variants (CNVs) in large population based samples are providing researchers with large data sets containing information on millions of genomic variations for thousands of individuals (1,2). Genome-wide association studies (GWAS) have gained increasing attention as it has become feasible and affordable to conduct studies involving thousands of samples and millions of variations per sample. Despite this windfall of data one of the major challenges of GWAS is to identify real causal variants and separate them from the millions of spurious variations, while also linking these variations to biological mechanism and disease pathogenesis by inference (3–10). To achieve this goal, researchers often need to browse through thousands of candidate SNPs, link these SNPs to genes or other functional genomic elements such as regulatory regions near these loci, and then familiarize themselves with the existing knowledge about the function and related phenomena and diseases linked to the SNPs, genes and other elements. These efforts, while necessary, are inefficient, and impractical for studies involving more than a handful of variations.
Varietas is a web-based database portal that has been designed to aid researchers to easily retrieve information on a set of variations (e.g. SNPs or CNVs), related genes and genomic elements in a batch like manner (Figure 1). The retrieved information can be explored using a web browser, or downloaded as a tab-delimited text file for further processing. Varietas also links out to several external resources that provide further information about the variations and genes of interest, such as the major genomic information resources Pubmed (11), dbSNP (11), SNPedia (12) and Ensembl (13). Varietas can be especially useful when used as a starting point for interpreting GWAS results, where the user can quickly enter a set of the top hits from the GWAS and easily get the fundamental information about these variations, related genes, diseases, and follow links to further external resources. Special consideration has been placed on keeping the user interface very simple, while still enabling users to have necessary control over the database queries. A major design feature is the ease of use such that no programming experience is needed to access and utilize Varietas.
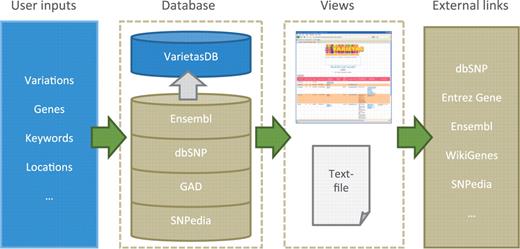
Overview of Varietas. Users can enter variety of different features such as SNPs, genes, keywords or locations, or any combination of them. These inputs are queried against VarietasDB that contains integrated data from various biological databases. Users can browse through the results using the web user-interface or download them as a tab-delimited text file. Links to external databases and resources are also provided for further exploration.
Description of the database
Data integration
Varietas integrates data from and links out to various SNP and genome databases and resources. Data is currently integrated from the following resources: Ensembl genome database, NCBI dbSNP database, The Genomic Association Database (GAD) (14) and SNPedia. These resources themselves integrate data from other resources. For example, disease data from Online Mendelian Inheritance in Man (OMIM) (15) and gene information from WikiGenes (16) are included through GAD and Ensembl, respectively. Query results from Varietas contain links to external resources such as NCBI dbSNP, NCBI Pubmed, NCBI Entrez Gene, Ensembl, WikiGenes and SNPedia.
Data is periodically integrated through extractors that retrieve data from the respective data sources, and then integrate and store the data in a relational MySQL database called VarietasDB. Variation information is primarily indexed and stored based on their dbSNP rs-numbers, allowing for other types of identifiers for variations that do not have assigned rs-number. Gene information and gene related information such as OMIM disease information is indexed and stored based on Ensembl gene identifiers and linked to variations using SNP–gene relationships from Ensembl, including information about the relationships such as SNPs relative location (e.g. exon, intron, downstream) and consequence (e.g. non-synonymous coding) to the gene.
If a single variation is linked to multiple data entries of the same type, e.g. consequence, phenotype or gene, queries will return a result set consisting of multiple rows indexed by the variation identifier and differing by the field(s) containing multiple entries (e.g. querying a SNP that is located within two individual genes will return two rows that contain the same variation information but differ in their gene information fields). In situations where external data sources contain dissimilar information for a variation (e.g. related phenotypes or linked genes) all available information is still indexed and available in the database. Users have the possibility to inspect the data to determine if the information is conflicting and what data sources are most reliable.
Information about the resource versions and extraction dates are available for Varietas users in order to track information such as version of genome assemblies and data builds. Varietas also archives and keeps online old versions of the integrated VarietasDB and web user interfaces, enabling reproducible research and tracking of data changes between versions.
User interface
Varietas’ web user interface (UI) has been developed to present users with a very simple to use yet powerful tool (Figure 2). UI consists of two main parts: basic and advanced search pages. Basic search provides users with all of the main functionality of Varietas while advanced search provides users with fine-tuning parameters for queries and returned results (e.g. what fields to retrieve and how the results are displayed). The main functionality of Varietas is to enter a batch of SNPs, genes, locations or keywords, and retrieve linked genomic variations, genes and related information such as gene and SNP descriptions and information about linked diseases and publications. Results are provided to users as a table that includes links to external resources. Results can also be downloaded as a tab-delimited text file for further processing with the users favorite spreadsheet software and bioinformatics tools. The web UI has been implemented using PHP and JavaScript programming languages.
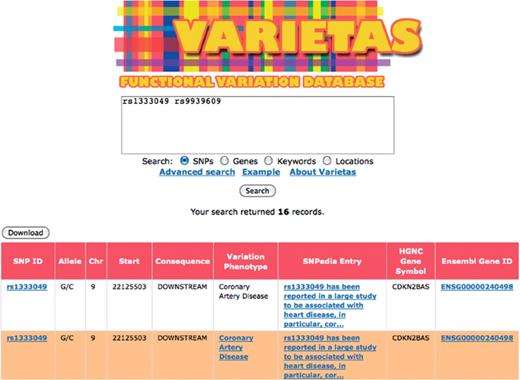
Screenshot of Varietas’ user interface showing partial results for basic query for a set of SNPs. Queries can be performed based on given set of variations, genes, keywords or genomic locations. Links in the results table can be followed to external information resources.
Discussion
Various resources for SNP information retrieval and annotation exist, and they have been compared in detailed reviews (17,18). When comparing Varietas to existing resources, Varietas adds new functionalities, improves existing ones and provides these services through a very simple and friendly UI that does not require specialized bioinformatics or programming skills from the users. When compared to existing genotype/phenotype databases such as SNPedia, dbGap (19), HGVbaseG2P (20) and similar databases (21) Varietas also provides information about SNPs that are not yet identified in GWAS studies, as well as information about linked genes and their phenotypes making it possible to predict novel phenotypic information for the variations. New and improved functionalities over existing tools include batch querying information from resources that do not have direct batch querying options (e.g. SNPedia), possibility to retrieve both combined SNP and gene information with a single query instead of having to combine multiple queries and the possibility to combine query parameters such as SNP and gene identifiers to free keywords that can include disease terms, gene descriptions and SNPedia entries. These findings can then be further examined with more comprehensive genetic association and disease resources such as HuGE Navigator (22) and OMIM.
The main strengths of Varietas are the easy to use web-based UI and the possibility to process large sets of SNPs to retrieve fundamental information about these SNPs, related genes and diseases. These results are gathered from sources that do not themselves allow batch queries. Integrating data from SNPedia, NHGRI GWAS Catalog (23) and The European Genome-phenome Archive (EGA) through Ensembl allows users to find focused information for previously characterized individual SNPs, while integrated gene information allows making new hypotheses about the SNP functions based on SNPs relations to genes, functions of those genes and related diseases.
One of the more useful new applications for Varietas is to use it to easily convert SNPs to gene sets, which can then be used for pathway and enrichment analysis using the wide variety of tools created for this purpose, such as Gene Set Enrichment Analysis (GSEA) (24).
Conclusions
Varietas is a novel SNP database resource for researchers working with genomic variation data sets or genome variation studies. Varietas includes a very simple and easy to use web-application that can be used to retrieve information about SNPs, related genes and diseases, based on data integrated from various genomic databases. In our own research projects Varietas has proved to be an excellent starting point when beginning to interpret results from analysis of high-throughput genotype data, such as GWAS. Based on our experience, we believe that Varietas can be useful for many other types of research as well. Varietas enables users to quickly browse through large numbers of SNPs and provides links to external resources for further information retrieval, and can be very useful for researchers working with GWAS and other variation data.
Several new data sources are planned to be integrated to Varietas in the future. We believe that when even greater volumes of genomic variation data becomes available, and our understanding of the links between genotypes and phenotypes improves through next-generation sequencing and large population based projects such as HapMap (2) and the 1000 Genomes Project (25), the need for tools like Varietas will be essential.
Funding
Finnish Graduate School of Molecular Medicine (to J.P.), and the Saastamoinen Foundation (to J.P. and G.W.). Funding for open access charge: University of Eastern Finland.
Conflict of interest. None declared.
Acknowledgements
The authors would like to thank Mitja Kurki and Petri Pehkonen for helpful comments during the design and implementation of this work.

{kind=link}
{kind=link}