





Abstract
Antibiotic resistance (AR) is a major global public health threat but few resources exist that catalog AR genes outside of a clinical context. Current AR sequence databases are assembled almost exclusively from genomic sequences derived from clinical bacterial isolates and thus do not include many microbial sequences derived from environmental samples that confer resistance in functional metagenomic studies. These environmental metagenomic sequences often show little or no similarity to AR sequences from clinical isolates using standard classification criteria. In addition, existing AR databases provide no information about flanking sequences containing regulatory or mobile genetic elements. To help address this issue, we created an annotated database of DNA and protein sequences derived exclusively from environmental metagenomic sequences showing AR in laboratory experiments. Our Functional Antibiotic Resistant Metagenomic Element (FARME) database is a compilation of publically available DNA sequences and predicted protein sequences conferring AR as well as regulatory elements, mobile genetic elements and predicted proteins flanking antibiotic resistant genes. FARME is the first database to focus on functional metagenomic AR gene elements and provides a resource to better understand AR in the 99% of bacteria which cannot be cultured and the relationship between environmental AR sequences and antibiotic resistant genes derived from cultured isolates.
Database URL:http://staff.washington.edu/jwallace/farme
Introduction
Antibiotic resistance (AR) is a significant and growing public health risk with bacterial mutations and horizontal gene transfer increasingly compromising the efficacy of antibiotic drugs. The Center for Disease Control and Prevention recently issued a report describing AR as a ‘serious threat’ to public health and estimating that at least 2 million people acquire antibiotic resistant infections each year (1). One of the four core actions highlighted in this report to prevent AR was tracking resistant bacteria; to do so requires consideration of both clinical and nonclinical (i.e. environmental) exposure pathways (2). In the environmental exposure pathway, antibiotic resistant bacteria are released into the environment through sewage, treated wastewater, agricultural run-off and other methods where they act as reservoirs for antibiotic resistant genes (3, 4). These reservoirs encourage the dissemination of antibiotic resistant genes between nonpathogenic and pathogenic bacteria via horizontal gene transfer (5, 6). In a recent review article, Bush et al. propose that controlling and preventing AR begins with understanding the development of AR in microorganisms found in the environment (7).
AR and the associated public health impacts have traditionally been studied using PCR and culture-based techniques. Because only a small fraction of microorganisms are estimated to be culturable (8, 9), these traditional methods misrepresent the AR potential of environmental microbial communities. Metagenomics has been shown to be a valuable approach for studying the prevalence of AR genes in the environment (10) and in contrast with PCR and culture based methods, allows for the characterization of the genetics of an entire microbial community. Metagenomic AR applications can be either sequence-based or functional, but both rely on DNA sequencing technologies. Sequence-based metagenomics involves the extraction and random sequencing of DNA directly from environmental media (e.g. water, soil, air etc.). Sequences are then compared with reference databases to assign taxonomic identity and functional potential. A diverse group of sequence-based metagenomic datasets are publicly available making global comparisons and analyses possible (11). In terms of AR, this extensive data has allowed for the development of a framework that integrates metagenomics into environmental AR risk assessment (12). However, without a laboratory-based confirmation of AR, it is not possible to determine functional AR through sequence-based metagenomics alone. Functional metagenomics similarly involves extracting DNA from environmental samples, but this metagenomic DNA is then cloned and expressed in a surrogate host (e.g. Escherichia coli) and screened for enzymatic activities such as AR (13). Resistant clones can then be selected and sequenced to identify nucleotide sequences conferring resistance, providing experimental validation of functional resistance and identification of novel ARGs (10). Despite the many studies that have used this valuable approach, there are currently no databases compiling functional metagenomic AR genes.
Current AR databases include the Antibiotic Resistance Genes Database (ARDB) (14) and the Comprehensive Antibiotic Resistance Database (CARD) (15). ARDB is an online tool that aims to provide a centralized resource to facilitate consistent characterization and identification of AR genes and contains ∼3000 non-redundant AR genes (14). More recently, CARD was released with ∼2200 non-redundant antibiotic resistant gene sequences (15). Both databases are assembled almost exclusively from genomic sequences derived from clinical bacterial isolates and include few, if any, functional metagenomic sequences.
In this article, we present the Functional Antibiotic Resistance Metagenomic Element (FARME) Database. FARME is the first database to focus on functional metagenomic AR gene elements rather than on individual antibiotic resistant genes derived from cultured clinical isolates. We have produced this database by compiling publically available DNA sequences from 20 functional metagenomics projects and their corresponding predicted protein sequences conferring AR. FARME also includes regulatory elements, mobile genetic elements and predicted proteins flanking AR genes. These features have been shown to be conserved between functional metagenomic AR sequences found in soil biomes and pathogenic clinical isolate sequences (16).
We have augmented authors’ GenBank (17) annotations with BLAST analysis of DNA and predicted proteins by searching current GenBank non-redundant protein and DNA sequence repositories and annotated conserved protein domains using hidden Markov model (HMM) analysis with the Pfam (18) and Resfams (19) HMM databases. HMM analysis serves as a valuable complement to traditional local similarity searching revealing highly specific AR protein motif conservation suitable for high-resolution analysis and visualization (19).
In addition, a FARME DB website dashboard (http://staff.washington.edu/jwallace/farme) was created to provide interactive evaluation and visualization of FARME AR elements by AR category, biome type and geographic location.
Database Construction and Content
GenBank and MetaGeneMark annotation
Functional metagenomic DNA sequences and annotations from 20 individual functional metagenomics projects (16, 29, 38) were downloaded from GenBank (17) along with their protein sequence predictions and annotations (if available). Results were loaded into MySQL tables created for DNA sequences, protein sequences and HMM predictions (Figure 1).
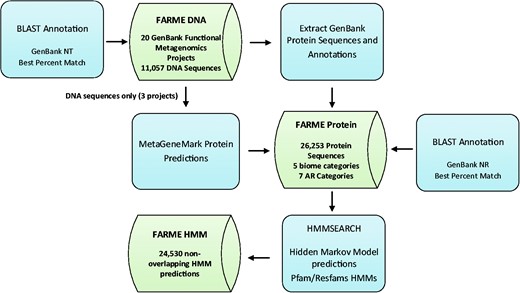
FARME database workflow showing analysis software components in blue and main database tables in green.
DNA sequences for three projects without corresponding protein sequence predictions (16, 29, 38) were analyzed with MetaGeneMark software (39) using default settings and predicted proteins were included in the FARME protein sequence table. Only sequences from uncultured metagenomic sources were considered excluding sequences derived from ‘mixed’ culture isolates.
GenBank and HMM sequence analysis annotation
Sequences were searched against the August 2015 GenBank non-redundant DNA and protein sequence databases using BLAST software (40) in order to annotate the best GenBank match with a minimum e-value threshold of 10−5 and excluding self-matches. The AR category for each DNA-sequenced clone was assigned using data from the methods section for each project.
Version 29 of the Pfam protein families database (18) and version 1.1 of the Resfams (19) AR HMM database were searched against the FARME predicted protein sequences FASTA file using the HMMSEARCH module of the HMMER version 3.1B2 software package (41) and the ‘trusted cutoff’ threshold. ‘Trusted cutoff’ is the score of the lowest scoring known true positive included in a full HMM profile alignment (42). Overlapping HMMs within the same protein region were resolved by choosing the HMM model with the highest HMMSEARCH bitscore for annotation. Non-overlapping HMMs within a predicted protein were annotated in the FARME HMM table along with the predicted gene sequence and position within the predicted gene. HMMs were designated as resistance elements for FARME website visualization based on their annotations in Pfam and Resfams databases. AR elements were partitioned into AR genes, transcriptional regulators and mobile genetic elements.
Web Framework
The FARME database website is built on a ‘LAMP’ (Linux, Apache, MySQL and PHP) open-source architecture. PHP scripts query MySQL tables returning JSON format data to Google Charts with Google Maps using AJAX (Asynchronous Javascript and XML) and HTML for geographical representation and drilldown into FARME projects as shown in Figure 2. Google map markers represent geographical sample collection coordinates, if available. Otherwise, markers represent project laboratory site coordinates.
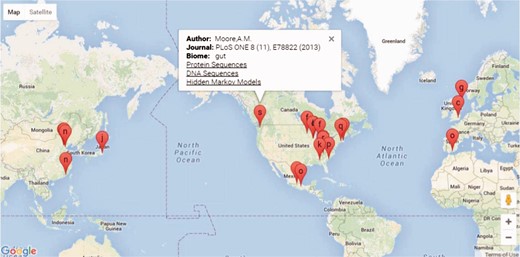
Screen shot of the FARME website project drill down feature showing geographical location of projects, if available, or project experiment sites with links to protein and DNA sequences and HMMs.
The HMM visualization browser tool (Figure 3) uses custom Pfam Javascript libraries (http://pfam.xfam.org). DNA and protein browser table entries link to their respective NCBI GenBank records. The complete set of records for each project are loaded into the Google Charts interface featuring sortable individual table fields and searching using native browser search features. We have also provided a BLAST interface (40, 43) as part of the web infrastructure to allow user-provided DNA or protein sequences to be searched against the FARME DB programs. Compatible web browsers tested include Apple Safari, Microsoft Edge, Microsoft Internet Explorer, Google Chrome and Mozilla Firefox.
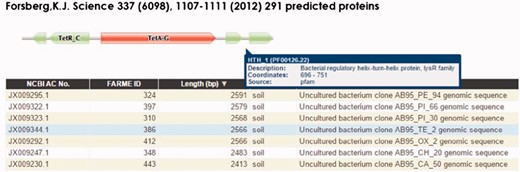
FARME website screen shot for a tetracycline resistant clone showing a tetracycline resistance HMM prediction flanked by TetR and LysR family transcriptional regulator HMMs. Users can interactively drill down into each sequence assembly for any of 20 FARME projects to help visualize genomic neighborhood HMM features including mouse over tooltips describing HMM feature details as shown above for LysR family transcriptional regulator.
Results
The FARME DNA table consists of 11,057 DNA sequences totaling 19,856,189 bases. The FARME protein table contains 26,253 corresponding predicted protein sequences with a total of 5,441,301 amino acids. The FARME HMM table is populated with 24,530 total non-overlapping HMM models and 2,250 different models found within 21,172 protein sequences. This includes 8,478 (35%) predicted AR HMMs, 1,369 (5.6%) transcriptional regulator HMMs and 360 (1.5%) mobile genetic element HMMs.
Figure 4 shows the AR categories present in FARME DB. Overall, functional AR gene elements including all predicted proteins were derived from two main biome types: soil and gut (fecal matter). Other biome types represented included wastewater treatment plants, oral and aquatic biomes.
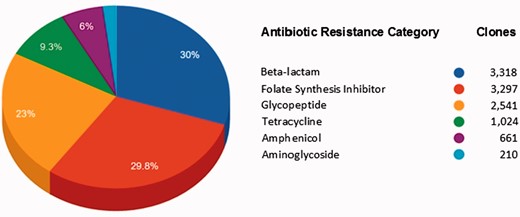
Main antibiotic categories used to select clones containing FARME DNA sequences for 20 individual projects.
Many FARME predicted proteins match known sequences in the GenBank protein sequence repository at a high percent identity: over 28% of FARME predicted proteins match sequences in GenBank at 100% identity and over two-thirds of FARME predicted genes (69%) match sequences in GenBank at >80% identity. Figure 5 shows the sequence similarity of 8,280 FARME predicted protein sequences containing AR resistance elements compared to GenBank and ARDB. FARME DB sequences have much higher similarity to GenBank sequences than to ARDB, thus illustrating the value of maintaining an up-to-date repository of functional metagenomic AR sequences not included in AR databases derived from cultured isolates. In addition, there are 1,334 FARME protein sequences with <80% similarity to both GenBank and ARDB suggesting potentially novel AR elements derived from environmental samples.
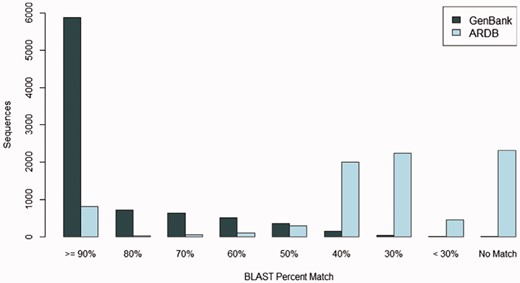
The number of FARME sequences within a percent identity bin from BLAST searching 8,280 FARME protein sequences containing AR resistance elements when compared with GenBank non-redundant protein and ARDB databases. FARME sequences show a dramatically higher percent identity with the GenBank non-redundant protein database than with the ARDB database illustrating the value of maintaining an up-to-date functional metagenomics AR database as a complement to clinically derived AR databases.
Functional antibiotic resistant metagenomics clone sequences with little or no identity to GenBank sequences included in FARME have been recently shown to contain genes which confer AR via new resistance models. Allen et al. (44) isolated beta-lactamase resistant clones from pristine Alaskan soil containing no known AR genes. They discovered a DNA binding response regulator gene with a 57% amino acid identity in GenBank enhancing resistance to the Beta-Lactam carbenicillin in E. coli. Forsberg et al. (45) used functional metagenomics clones isolated from multiple soil types to discover a family of tetracycline resistance genes dubbed ‘tetracycline destructases’. This new family of tetracycline genes shows no amino acid identity to known tetracycline resistance genes. However, tetracycline inactivating clones in this project share a common ResFams HMM motif, ‘TE Inactivator’, which is also contained within tetracycline resistant clones found in another FARME project which characterizes the pediatric gut resistome (29).
Discussion
FARME is the first database to focus on functional metagenomic AR genes and gene elements sourced from environmental samples rather than on individual antibiotic resistant genes derived from cultured clinical isolates. FARME contains over seven times the number of non-redundant protein sequences compared with other AR databases such as ARDB and CARD and includes essential information about the ‘genomic neighborhood’ proximal to functionally tested genes, e.g. well-known regulatory elements identified by HMM analysis (e.g. TetR, LysR) (16) (Figure 3). As such, FARME provides a basis for analyzing the sequence similarity between functional AR genes derived from the environment and future sequences from clinical and metagenomic studies. Although the majority of FARME sequences share similarity to known AR genes from clinical studies, a number of FARME sequences share little to no similarity to known AR genes, suggesting novel AR genes from the environment.
Four projects out of 20 in FARME utilize high-throughput second-generation DNA sequence technology (16, 24, 25, 29) and one of the newest projects (36) utilizes third-generation sequencing which combines high-throughput with long-read sequencing reads (>7 kb). In the future, next-generation sequencing (NGS) will provide greater throughput, lower cost and higher resolution for functional metagenomic sequencing experiments. Adoption of NGS will necessitate new search strategies providing maximum speed, sensitivity and specificity for gene annotation. As such, researchers utilizing NGS will require easy to use analysis frameworks and up-to-date databases like FARME to help achieve goals such as providing timely global AR surveillance. In addition, leveraging HMMs to identify specific AR genes and mechanisms of action within predicted protein sequences offers the potential of high-performance screening of thousands of metagenomic sequences in a fraction of the time taken by traditional similarity searching methods. Recent improvements in the Hidden-Markov model software HMMER, used to generate the FARME HMM table in our database, can provide search speeds four orders of magnitude faster than BLAST (46).
In summary, FARME is the first AR database to focus exclusively on environmentally derived metagenomic genes, and as such provides an opportunity for researchers to access and analyze AR genes found outside of the clinical setting. The database, annotation schema and browser interface provide a valuable and needed resource to better understand and characterize AR elements in the majority of uncultured bacteria and their genetic similarity to AR elements derived from cultured isolates.
Funding
This work was supported by the National Oceanic and Atmospheric Administration (NOAA)-funded Pacific Northwest Consortium for Pre- and Post-doctoral Traineeships in Oceans and Human Health [grant number S08-67883 MOD03] and was also supported by the National Science Foundation (NSF) (grant numbers 0910624 and 1128883). This publication was also made possible by United States Environmental Protection Agency (US EPA) grant 8357380. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NOAA, NSF or the US EPA. Further, US EPA does not endorse the purchase of any commercial products or services mentioned in the publication. This work was also supported by The University of Washington, Center for Exposures, Diseases, Genomics and Environment, of the National Institutes of Health under award number: P30ES007033.
Conflict of interest. None declared.
References
Author notes
Present Address: Jesse A. Port, Center for Ocean Solutions, Stanford University, Stanford, CA, USA.
Citation details: Wallace,J.C., Port,J.A., Smith,M.N. et al. FARME DB: a functional antibiotic resistance element database. Database (2016) Vol. 2016: article ID baw165; doi:10.1093/database/baw165

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}