





Abstract
Using bioinformatics, putative cis -regulatory sequences can be easily identified using pattern recognition programs on promoters of specific gene sets. The abundance of predicted cis -sequences is a major challenge to associate these sequences with a possible function in gene expression regulation. To identify a possible function of the predicted cis -sequences, a novel web tool designated ‘ in silico expression analysis’ was developed that correlates submitted cis -sequences with gene expression data from Arabidopsis thaliana . The web tool identifies the A. thaliana genes harbouring the sequence in a defined promoter region and compares the expression of these genes with microarray data. The result is a hierarchy of abiotic and biotic stress conditions to which these genes are most likely responsive. When testing the performance of the web tool, known cis -regulatory sequences were submitted to the ‘ in silico expression analysis’ resulting in the correct identification of the associated stress conditions. When using a recently identified novel elicitor-responsive sequence, a WT-box (CGACTTTT), the ‘ in silico expression analysis’ predicts that genes harbouring this sequence in their promoter are most likely Botrytis cinerea induced. Consistent with this prediction, the strongest induction of a reporter gene harbouring this sequence in the promoter is observed with B. cinerea in transgenic A. thaliana .
Database URL:http://www.pathoplant.de/expression_analysis.php .
Introduction
Eukaryotic gene expression is largely regulated by the binding of transcription factors (TFs) to cis -sequences in promoter regions. To identify cis -sequences that may have a specific regulatory function, pattern recognition programs can be used to identify common cis -sequences in promoters of a set of co-regulated genes ( 1 ). Such bioinformatic approaches have been used in recent years for the discovery of large numbers of conserved cis -regulatory sequences, many of which are not associated with known cis -sequences ( 2–6 ). The abundance of predicted cis -sequences represents a major challenge for defining their function in regulation of gene expression.
One way to identify the conditions upon which a cis -sequence may confer gene expression is to analyse if genes harbouring this sequence in their promoter show a specific expression profile under certain environmental conditions ( 7 ). This has been done for many sequences predicting known and novel expression profiles ( 8–10 ). In several cases, these sequences have also been tested experimentally ( 8 , 11 ). This shows that such an approach is a useful way to identify the possible function of specific cis -sequences. For such predictions, it may be particularly helpful to have an online web tool that permits such an analysis for any given cis -sequence. To facilitate such an analysis, a new web tool designated ‘ in silico expression analysis’ has been implemented in the PathoPlant database ( 12 , 13 ). The database is manually annotated with data from the literature. Currently, it contains data for 99 plant species and varieties, 107 pathogens and 638 molecules from 619 references ( 14 ). These data represent 350 interactions and 370 reactions. Via a recently developed function, molecules and reactions annotated within PathoPlant can be visualized as signalling pathway maps. A map of all reactions and molecules annotated to PathoPlant can be generated as well as specific pathway maps starting from a selected molecule ( 14 ). In addition, 144 different microarray data sets from Arabidopsis thaliana , corresponding to 36 different abiotic and biotic stimuli, have been annotated to PathoPlant.
The newly developed ‘ in silico expression analysis’ tool can be used to identify the biotic and abiotic stimuli that may induce or repress expression of genes harbouring a specific cis -sequence under investigation. Using the web tool, the potential cis -sequence can be submitted to perform a genome-wide A. thaliana promoter screening. The gene sets obtained are used to calculate mean induction factors for every A. thaliana microarray experiment stored within PathoPlant. A negative ‘induction factor’ would mean that these genes are downregulated. These mean values are normalized according to overall expression values of each stimulus. This results in a ranked list of microarray experiments according to their mean induction factors. The most probable stimuli to which genes harbouring the potential cis -element in their promoter are responsive to can be identified by looking at the highest-ranked stimuli for upregulated genes or the lowest ranked stimuli for downregulated genes. This article describes the implementation of the web tool and a proof of concept analysis with known cis -sequences that confer stress-responsive gene expression. In addition, using the recently identified WRKY70 TF binding site CGACTTTT ( 15 ) and reporter gene technology in transgenic A. thaliana , the prediction made by an ‘ in silico expression analysis’ was confirmed.
Methods
Microarray data
The PathoPlant database harbours microarray expression data primarily for biotic and abiotic stress conditions ( 5 , 13 ). Most of the microarray data were generated in the AtGenExpress project ( 7 ) and were downloaded from TAIR, NASCArrays, ArrayExpress and NCBI GEO ( 16–19 ). Microarray data were normalized using the Affymetrix MAS5 algorithm ( 20 ). Currently, 144 different microarray data sets corresponding to 36 different abiotic and biotic stimuli have been annotated to PathoPlant. All data sets, array type and a link to the expression set used for downloading the data can be found on the documentation page of PathoPlant at http://www.pathoplant.de/ . In addition to the 144 data sets for abiotic and biotic stimuli, two data sets correspond to inflorescence-specific gene expression. The data can be accessed through the ‘Microarray expression’ tool at http://www.pathoplant.de/ as described earlier ( 13 ).
The ‘ In silico expression analysis’ web tool
To bioinformatically assess the functionality of identified cis -sequences, a new web tool was developed that can be accessed online at http://www.pathoplant.de/expression_analysis.php . It provides a bioinformatic approach to investigate whether genes harbouring specific cis -sequences are responsive to certain biotic and abiotic stresses. The web tool validates the possible role of the cis -sequence to confer the identified stress-responsive gene expression. The tool uses microarray expression data from the PathoPlant database to calculate mean induction factors for gene sets that contain a submitted sequence within their promoters. Positive mean induction factors (>1) describe upregulated genes, and negative mean induction factors (<−1) describe downregulated genes. The statistical significance of the mean induction factors is assessed by calculating a P- value and by applying a false discovery rate (FDR) P -value adjustment. Such information is used to evaluate the probability of a given sequence to be a putatively functional cis -element.
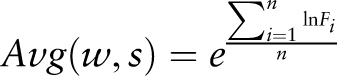
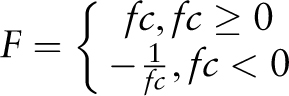
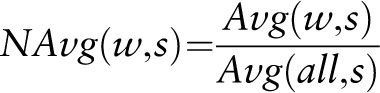
Statistical significance of the average mean expression values calculated for the different gene sets is assessed by means of a raw P- value calculation with a Student's t -test and subsequent FDR P -value adjustment.
The following data are used for raw P -value calculation for a given gene set that harbours the submitted cis -sequence within the selected promoter region in comparison with all genes present on the microarray chip: the average mean expression ( mean ), variance of the individual induction factors ( var ) and number of induction factors ( n ) used to calculate expression. These data are determined by the PathoPlant database server each time a new calculation is performed. The raw P -value is calculated by the Apache web server using PHP (version 5.3.11) and the stats_cdf_t function from the PECL stats extension package. This function accepts the t -value that is calculated from mean , var and n as parameters to return the raw P- value as observed significance associated with an one-tailed unpaired t -test. This compares the distribution of the individual induction factors obtained for the genes harbouring the submitted cis -sequence within the selected promoter region under each stress condition with the overall distribution of induction factors for all genes under the same stress condition. Because t -test and P -value calculation is performed on correlated data sets, Benjamini–Hochberg (BH) FDR P -value adjustment is applied to calculate BH-adjusted (FDR) P -values ( 23 ). By using standard PHP functions, the adjustment is performed from a sorted list of raw P -values and the number of data sets ( 24 ). These data are added to the output created by the in silico expression analysis tool, and in that way, it is possible to determine the significance value of a calculated average mean expression value for a given stress.
Once mean induction factors, raw and BH-adjusted (FDR) P- values are calculated, results are ordered according to mean induction factor values. The results are displayed online, and they can be resorted according to the calculated raw or BH-adjusted (FDR) P- values or according to the stimulus.
T-DNA constructs
The transfer DNA (T-DNA) construct for A. thaliana transformation was generated in the vector pGPTV_bar ( 25 ). For this, the cis -sequence was amplified by polymerase chain reaction (PCR) from a plasmid containing four copies of sequence 20 in pBT10-GUS ( 5 ) using primers 5′-TAGC AAGCTT GAATTCGGCGCGCCACTAGT-3′ and 5′-AT CCCGGG GGTGGCCACTCGAGC-3′. The PCR product was cut with HindIII and SmaI and cloned into the T-DNA vector digested with the same enzymes. After sequencing of the recombinant T-DNA vector, the resulting plasmid pSeq20_GPTV_bar was transformed into Agrobacterium tumefaciens C58C1 ( 26 ). The plasmid pSeq20_ GPTV_bar contains four copies of sequence 20 upstream of a minimal promoter (TATA-box) linked to the uid A reporter gene encoding β-glucuronidase (GUS). For all recombinant DNA, work standard protocols were used ( 27 ). Sequence analysis was done by GATC Biotech (Konstanz, Germany).
Transformation of A. thaliana
Arabidopsis thaliana accession Col-0 was transformed following the floral dip transformation protocol ( 28 ). For transformation, A. tumefaciens C58C1 harbouring plasmid pSeq20_GPTV_bar was used. After harvesting the seed of the transformed plants, transgenic plants were selected on medium containing 30 mg/l phosphinothricin. A total of 14 independent transformants were obtained. Segregation analysis of transgenic offspring in the T1 generation revealed that five lines harbour one T-DNA locus (lines 3, 6, 8, 13 and 14). These lines were subjected to pathogen infection and reporter gene assays.
Pathogen infection
All pathogen infections were performed with 5- to 8-week-old transgenic A. thaliana lines grown under short-day conditions (8 h light, 16 h dark). For infection with Botrytis cinerea (strain B05.10), the fungus was grown on potato dextrose agar (PDA) medium (Carl-Roth, Karlsruhe, Germany) at 25°C in 150-mm petri dishes. Infections were done according to Mengiste et al. ( 29 ). For infection, spores of a 10-day-old B. cinerea culture were recovered using 10–15 ml of Sabouraud maltose broth [(SMB), 40 g/l maltose, 10 g/l pepton, pH 5.6)]. Spores were scrabbed from the B. cinerea mycelium with a glass pipette. The spore suspension was recovered by filtration through gauze, and the spore concentration was determined using a haemocytometer. For infection, the suspension was adjusted to 1 × 10 5 spores/ml in SMB, and a 10-µl droplet of the spore suspension was applied to the A. thaliana leaves to be infected. To maintain high humidity, the plants were kept in closed containers saturated with water vapour. After 3 days, leaves were harvested for reporter gene assays.
For infection with Pseudomonas syringae pv. tomato DC3000, a virulent (containing the vector pVSP61) and an avirulent strain (containing the vector pVSP61 expressing avrRPM1 ) were used. Both strains were grown on king's B (KB) medium (20 g/l pepton, 1.5 g/l K 2 HPO 4 , 1.5 g/l MgSO 4 7H 2 O, 10 ml/l glycerol, 15 g/l agar) containing kanamycin (50 mg/l) and rifampicin (50 mg/l). After incubation for 2 days at 25°C, 5 ml of liquid KB medium (with antibiotics) was inoculated with a single colony and grown overnight at 25°C. Subsequently, 50 ml of prewarmed KB medium was inoculated with 1 ml from the over-night culture and incubated for another 5–6 h at 25°C. The cells were precipitated by centrifugation (3000 g , 10 min) and resuspended in sterile deionized water to a final optical density at 600 nm of 0.2. A 1:10 dilution of this suspension was used for leaf infiltration. For this, a needleless syringe containing 1 ml of the diluted bacterial suspension was used to infiltrate the abaxial side of a leaf by slightly pressing the syringe against the leaf. The infiltrated area was usually 45 mm in diameter. As a control, water-only infiltrations were performed in the same way. The plants were kept in closed containers for 2 days and subsequently subjected to reporter gene assays.
Reporter gene assays
After pathogen infection on single leaves of A. thaliana , single infected and single non-infected or water-inoculated leaves were subjected to quantitative reporter gene (GUS) assays ( 30 ). For this, single leaves were homogenized in liquid nitrogen. Two hundred microliters of GUS extraction buffer (50 mM NaPO 4 , pH 7, 10 mM Na 2 EDTA, 0.1% Triton X-100, 0.1% N -laurylsarcosine, 10 mM β-mercaptoethanol) was added, mixed, and the cell debris was precipitated by centrifugation (10 min, 16 000 g , 4°C). The supernatant was recovered, and the protein concentration was determined according to Bradford ( 31 ). The protein concentration was adjusted to 80 µg/ml using GUS extraction buffer. Twenty-five microliters of the diluted protein extract (2 µg) was transferred into a well of a black 96-well microtiter plate. A total of 225 µl of GUS reaction buffer (50 mM NaPO 4 , pH 7.0, 10 mM Na 2 EDTA, 0.1% Triton X-100, 0.1% N -laurylsarcosine, 10 mM β-mercaptoethanol, 1 mM 4-methylumbelliferyl-β- D -glucuronide) was added, and the plate was inserted into a TriStar LB 941 microplate reader (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany) and incubated at 37°C for 10 min before measurements at 37°C. For continuous measurement of GUS activity, the samples in each well were then measured every 15 min for 1 s for the next 3 h (excitation 360 nm, emission 460 nm). Afterwards, for each well, a linear regression for the time period with a linear increase of fluorescence was performed. Non-linear parts were excluded from the regression. The slope of the regression line was then transformed into pmol 4-MU min −1 mg −1 . For this, a calibration of fluorescence units with defined amounts of 4-MU was performed in the TriStar. The results shown here are mean values from two ( B. cinerea ) or three ( P. syringae strains) independent experiments with two replicates each. Error bars represent standard deviations. The induction factors are calculated from the mean values mentioned above.
Results
The ‘ In silico expression analysis’ online web tool
An ‘ in silico expression analysis’ can be performed online to validate short DNA sequences as cis -regulatory sequences potentially conferring gene expression to specific biotic and abiotic stress conditions. Figure 1 shows a screen shot of the online tool with the result obtained with the ‘Demo’ sequence. When selecting ‘Demo’, the sequence TACCGACAT appears as the input sequence. This sequence was first identified in the promoter of the drought-responsive RD29A gene from A. thaliana and was demonstrated to function as a cis -acting element involved in the induction of RD29A expression by low-temperature stress ( 32 ). The cis -sequence submitted is used by the web tool to perform a genome-wide A. thaliana promoter screening. A 250-, 500- (default) or a 1000-nt-long upstream region can be selected for analysis. There is an option to exclude genes potentially regulated by small RNAs or microRNAs (miRNAs) from the analysis. These were identified in the A. thaliana genome and annotated to the AthaMap database, which is linked to PathoPlant ( 13 , 33 , 34 ). In all, 55 genes harbour the ‘Demo’ sequence in the selected 500-nt upstream region ( Figure 1 ). The gene set harbouring the submitted cis -sequence in the selected promoter region is used to calculate mean induction factors for every A. thaliana microarray experiment stored within the PathoPlant database. These mean values are normalized according to overall expression values of each stimulus. This results in a ranked list of microarray experiments according to their mean induction factors. Consistent with its known function, the result with the ‘Demo’ sequence reveals this sequence to be associated with genes responding to cold stress ( Figure 1 ). The mean induction factor of the genes harbouring this sequence in the selected promoter region is 2.87 for the microarray ‘cold-stressed shoots 24 hr’, followed by four other microarrays for which lower mean induction factors of the genes were determined under cold stress. For each stimulus, the number of expression values used for mean induction factor calculation is given. The corresponding P- value is also calculated for each mean factor to assess its significance. The number of expression values, here 74, is linked to a new window. Figure 2 shows a partial screen shot of this window revealing that 37 (‘number of genes’) of the 55 genes are actually on the microarray ‘Cold-stressed shoots 24 hr’ (‘stimulus’). Further information given on this page includes the gene identifier (‘Gene’), the orientation and position of the cis -sequence relative to the gene start and the induction factor of each experiment, as well as the mean induction factor of the gene ( Figure 2 ). The orientation and relative distance refers to the distance of the first match position to the point of reference that can either be TSS, if known, or the translation start codon, if the TSS is unknown. The individual and mean induction factors of each gene, as well as the number of replicates ( n ) and the base-10 logarithm of the standard deviation for mean induction factor calculation of each gene, are shown. By default, the result tables are ranked by mean induction factors ( Figures 1 and 2 ) and can be resorted in descending or ascending order by selecting the headers of the tables. By selecting the number of genes (55, Figure 1 ), a gene list is shown in a table (not shown). In this gene list, gene descriptions are displayed when selecting the arrow next to the table header ‘Gene’. The list of genes obtained by the ‘ in silico expression analysis’ can be submitted directly to the ‘Microarray expression’ function of PathoPlant to obtain expression data of these genes for all stimuli. In addition, this list can also be transferred to AthaMap’s gene analysis function for a TF binding site analysis ( 35 ).
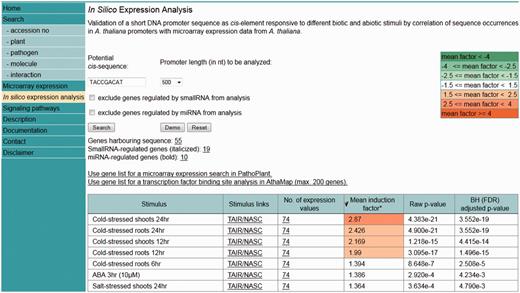
Screenshot of the ‘in silico expression analysis’ web tool showing the result obtained with the ‘Demo’ sequence.
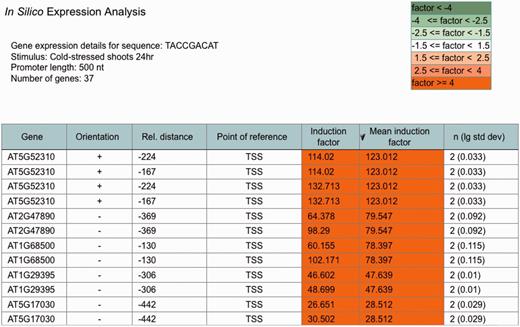
Partial screenshot showing the most highly cold-induced genes identified with the ‘Demo’ sequence. The table identifies the individual genes obtained in the ‘ in silico expression analysis’ for the selected sequence and the selected stress. Furthermore, it shows the orientation and relative distance of the sequence to the point of reference (TSS) in each gene. The induction factor of each replicate, the mean induction factor and the number of replicates ( n ) is displayed. The table is sorted according to mean induction factor.
Validation of the ‘ in silico expression analysis’ web tool with known cis -regulatory sequences
To test the performance of the ‘ in silico expression analysis’ web tool, additional cis -sequences associated with stress-specific gene expression were investigated in addition to the ‘Demo’ sequence used in Figure 1 . Figure 3 shows the cis -sequences and their predicted stress responsiveness. When the sequence CACGTGTC is submitted using the ‘ in silico expression analysis’ web tool with default settings, the genes harbouring this sequence within their promoter were found to be most strongly upregulated in the microarray expression data set abscisic acid ( Figure 3 A). This sequence has previously been associated with abscisic acid-responsive genes ( 2 , 9 ).
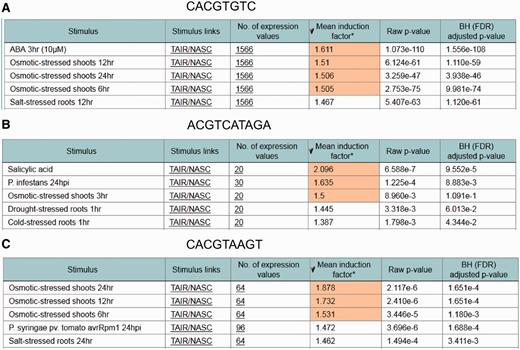
Examples for identifying stress responsive cis -elements using the in silico expression analysis web tool. In each case, the cis -sequence used for in silico expression analysis with default settings is shown together with the five microarray expression data sets for which the most significant correlation between occurrence of the cis -sequence within the promoter and the expression of the associated genes was detected. Cis -sequences are shown. ( A ) An abscisic acid response element. ( B ) A salicylic acid response element. ( C ) A dehydration and senescence response element.
The sequence ACGTCATAGA was previously associated with salicylic acid-responsive genes ( 36 ). This sequence is part of LS7, a regulatory sequence from the pathogenesis-related gene PR1 , which is upregulated by salicylic acid. Figure 3 B shows that the microarray expression data set in which genes harbouring this sequence in their promoter were found to be upregulated most strongly is salicylic acid.
Another example is the dehydration and senescence-responsive sequence CACGTAAGT from the SAG113 promoter ( 37 ). Figure 3 C shows that genes harbouring this sequence in their promoter were found to be upregulated most strongly by osmotic stress, which causes dehydration.
These examples show that previously identified cis -sequences associated with specific stress conditions can be used in the ‘ in silico expression analysis’ to identify the correct corresponding microarray data sets.
Prediction of stress conditions for genes harbouring a novel cis -regulatory sequence
Recently, a novel elicitor-responsive cis -regulatory sequence from A. thaliana , CGACTTTT, was predicted bioinformatically and was shown to be a binding site for the WRKY70 TF ( 5 , 15 ). Although the cis -sequence, designated WT-box and bound by WRKY70, is enriched in promoters of genes upregulated in a WRKY70 overexpression line, the primary stimulus to which genes harbouring this sequence in their promoter respond to was unknown.
To identify the pathogenic stimulus or any other stress condition that most likely induces genes harbouring the new cis -sequence CGACTTTT, an ‘ in silico expression analysis’ was performed. The analysis was done by submitting the sequence CGACTTTT using default settings of the web tool, except for the small RNA-regulated genes. These were excluded from the analysis because these genes are most likely also post-transcriptionally regulated. Figure 4 A shows that 355 genes harbour the cis -sequence within a 500-nt upstream region. ‘ In silico expression analysis’ indicates that genes harbouring this sequence in their promoter are most likely responsive to B. cinerea ( Figure 4 A). The mean induction factor for the genes on the microarray data set was 1.245 ( B. cinerea ), which is low, but the low P -value (1.4E-9) for B. cinerea may indicate a significant correlation of these genes with their induction by B. cinerea .
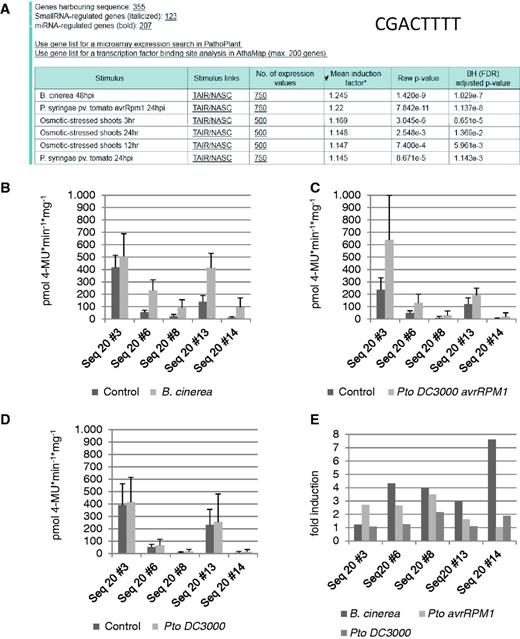
‘ In silico expression analysis’ and experimental validation of the cis -regulatory sequence CGACTTTT. ( A ) The in silico expression analysis result with sequence CGACTTTT. ( B–D ) Quantitative GUS expression (pmol 4-MU min −1 mg −1 ) after infection of transgenic A. thaliana lines with B. cinerea (B), P. syringae pv . tomato avrRPM1 (C) and P. syringae pv. tomato (D) compared with the uninfected control. ( E ) The fold induction determined from the change between the GUS values of uninfected and infected plants.
Experimental verification of predicted stress conditions for the cis -regulatory sequence CGACTTTT
To test the predictions of the ‘ in silico expression analysis’ experimentally, five independent transgenic promoter-reporter gene lines were analysed. These lines harbour four copies of the cis -sequence, containing the sequence CGACTTTT upstream of a minimal promoter and the uidA (GUS) reporter gene (Methods).
When these transgenic lines are subjected to infection with spores of B. cinerea , all lines show upregulation of the reporter gene compared with the uninfected control ( Figure 4 B). When the same transgenic lines are subjected to infiltration with P. syringae pv. tomato avrRpm1 , these lines also show upregulation of the reporter gene compared with the uninfected control ( Figure 4 C). When the same transgenic lines are subjected to infiltration with P. syringae pv. tomato , these lines do not show upregulation of the reporter gene compared with the uninfected control ( Figure 4 D). As a negative control, a transgenic line containing only the minimal promoter without the cis -sequence upstream of the uidA reporter gene was also tested with and without pathogen infection. Reporter gene expression values were always <15 pmol 4-MU min −1 mg −1 (not shown).
When comparing the induction factors of transgenic lines obtained with the three different pathogens, four of the five lines correspond to the prediction made by the ‘ in silico expression analysis’ because induction is strongest (3–7.6-fold) after B. cinerea infection (lines 6, 8, 13 and 14, Figure 4 E). In contrast, lines 6, 8 and 13 show a lower mean induction (1.6–3.5-fold) by P. syringae pv. tomato avrRpm1 . The induction factor for P. syringae pv. tomato is mostly around 1, which means that no reporter gene induction can be detected.
Discussion
Using pattern recognition programs on promoter sequences of specific gene groups, it is fairly straightforward to identify conserved sequence motifs ( 1 ). In contrast to this, the functional analysis of these cis -sequences is more elaborate. If conserved sequence motifs were established from co-regulated gene groups, normally the conditions under which the genes are co-regulated are known and may be used to assess whether the sequences identified in the promoters of these genes confer corresponding reporter gene activity ( 5 ). However, often only the genomic distribution of short sequences was established, and the functional significance of specific sequences was mainly deduced from known cis -sequences ( 22 ). In such a study, 65 536 different 8-nt-long sequences, so-called words, were investigated with respect to frequency and positional distribution in the A. thaliana genome ( 9 ). This study classified the 8-nt-long sequences using gene expression information. Therefore, cis -sequences that occur in promoters were associated with specific gene expression profiles. For example, the abscisic acid response element, CACGTGTC, when found in a 1000-bp upstream region strongly correlated with induction by 10 µM abscisic acid ( 9 ). This sequence was used as one of the ‘proof of concept’ sequences for the ‘ in silico expression analysis’ web tool showing the previously determined association with abscisic acid-response genes ( Figure 3 A). This demonstrates that the ‘ in silico expression analysis’ web tool, which permits the submission of any putative cis -sequence for analysis with respect to gene expression data, will be useful. Although several known cis -sequences that were known to be associated with specific expression profiles were correctly identified with the associated microarray data set, the tool has certain limitations. If a cis -sequence is too short, the number of genes harbouring the sequence in the promoter will exceed the capacity of the system. If such a sequence is submitted, the web tool will display the statement: ‘The number of genes containing the sequence is too high. Please try a larger sequence or a shorter promoter length’. A more general problem when submitting a cis -sequence will be that no stress conditions are identified with the microarrays in the database. This is reminiscent of an earlier analysis in which no stress conditions could be associated with specific cis -sequences, although the cis -sequences were overrepresented in promoters of genes upregulated in a specific microarray data set ( 8 ). This may be due to combinatorial control of gene expression, requiring a second cis -sequence for specifying a specific gene expression profile ( 38 , 39 ). Such combinatorial control of gene expression is known for many cis -sequences and their binding TF ( 40 , 41 ).
In the work presented here, the ‘ in silico expression analysis’ web tool was successfully used to determine the biotic stress response conditions for genes harbouring a novel cis -regulatory sequence designated WT-box ( 15 ). This sequence, CGACTTTT, was detected when pattern recognition programs were used on promoters of A. thaliana genes upregulated by pathogenic stimuli ( 5 ). The reverse complement sequence AAAAGTC was previously detected to be enriched in promoters of genes responsive to flagellin 22, NPP1 and P. infestans ( 4 ). The sequence CGACTTTT was shown to be bound by WRKY70, extending the range of known WRKY binding sites ( 15 , 42 ). The ‘ in silico expression analysis’ performed with this sequence predicted that genes harbouring this sequence are most likely upregulated by B. cinerea ( Figure 4 A). Consistent with this proposal, four of five transgenic A. thaliana lines harbouring a reporter gene construct with synthetic promoters containing four copies of this sequence show the most prominent induction after B. cinerea infection ( Figure 4 B). This may indicate that genes harbouring this cis -sequence in their promoter may play a role during B. cinerea infection. Because this sequence is bound by WRKY70, it is interesting that a wrky70 mutant is more sensitive to B. cinerea infection ( 43 ). This may indicate that B. cinerea -responsive genes are no longer upregulated in the mutant.
In summary, the work presented here shows that the ‘ in silico expression analysis’ web tool can be used to predict stress conditions that are most likely inducing genes harbouring a specific cis -sequence in their promoter region.
Acknowledgements
We would like to thank Paul Tudzynski (Universität Münster) for the B. cinerea strain and Jane Parker (MPI Köln) for P. syringae strains. We are grateful to the anonymous reviewers for their help improving the presentation and functionality of the web site.
Funding
The Federal Ministry for Education and Research (BMBF), Germany and the KWS SAAT AG, Einbeck, Germany. Funding for open access charge: Technische Universität Braunschweig.
Conflict of interest . None declared.
References
Author notes
Citation details: Bolívar, J.C., Machens, F., Brill, Y., et al. ‘ In silico expression analysis’, a novel PathoPlant web tool to identify abiotic and biotic stress conditions associated with specific cis -regulatory sequences. Database (2014) Vol. 2014: article ID bau030; doi:10.1093/database/bau030

{kind=link}
{kind=link}
{kind=link}
{kind=link}