





Abstract
With the advent of high-throughput sequencing technologies, many staphylococcal genomes have been sequenced. Comparative analysis of these strains will provide better understanding of their biology, phylogeny, virulence and taxonomy, which may contribute to better management of diseases caused by staphylococcal pathogens. We developed StaphyloBase with the goal of having a one-stop genomic resource platform for the scientific community to access, retrieve, download, browse, search, visualize and analyse the staphylococcal genomic data and annotations. We anticipate this resource platform will facilitate the analysis of staphylococcal genomic data, particularly in comparative analyses. StaphyloBase currently has a collection of 754 032 protein-coding sequences (CDSs), 19 258 rRNAs and 15 965 tRNAs from 292 genomes of different staphylococcal species. Information about these features is also included, such as putative functions, subcellular localizations and gene/protein sequences. Our web implementation supports diverse query types and the exploration of CDS- and RNA-type information in detail using an AJAX-based real-time search system. JBrowse has also been incorporated to allow rapid and seamless browsing of staphylococcal genomes. The Pairwise Genome Comparison tool is designed for comparative genomic analysis, for example, to reveal the relationships between two user-defined staphylococcal genomes. A newly designed Pathogenomics Profiling Tool (PathoProT) is also included in this platform to facilitate comparative pathogenomics analysis of staphylococcal strains. In conclusion, StaphyloBase offers access to a range of staphylococcal genomic resources as well as analysis tools for comparative analyses.
Database URL: http://staphylococcus.um.edu.my/
Introduction
The genus Staphylococcus in the bacterial family Staphylococcaceae is a common bacterial genus that is widely distributed throughout the world. Some known Staphylococcus species are part of the natural fauna present on the body and can be found on mucus membranes and skin. Staphylococci are facultative anaerobic gram-positive spherical bacteria that occur in microscopic clusters resembling grapes. Staphylococcal pathogens are resistant to many antibiotics, forcing researchers to find better ways of fighting these pathogens. Staphylococcus aureus and Staphylococcus epidermidis are the two most characterized and studied staphylococcal bacteria, and S. aureus is a significant human pathogen worldwide. It is reported that one-fifth of the human population are long-term carriers of S. aureus (1). This bacterial species forms biofilms on medical devices, causing pneumonia, osteomyelitis, meningitis, endocarditis and septicaemia. In biofilms, the cells will be held together and exhibit altered phenotype with respect to bacterial metabolism, physiology and gene transcription (2). This pathogen has gained significant attention because of its multi-drug resistance in methicillin-resistant S. aureus (3) and vancomycin-resistant S. aureus (4), which have made this pathogen difficult to combat.
Recently, many genomes of staphylococcal bacteria have been sequenced by many laboratories, including research, public health and clinical laboratories, using high-throughput sequencing technologies (5–10). The availability of these genome sequences from different sources has made it possible to perform genome-wide comparative analyses. Such comparative analysis will have a profound impact on understanding the biology, diversity, evolution and virulence of the staphylococcal bacteria, which may be useful in successfully combatting the staphylococcal pathogens.
To facilitate this area of research, a specialized database system for Staphylococcus is necessary for the storage of the dramatically increasing genomic data of staphylococcal bacteria, to present the data in a manner that is easy to access and useful, and to enable the analysis of these genomic data, particularly in the field of comparative genomics. Here, we present StaphyloBase, a staphylococcal genomic resource platform powered by advanced web technologies and in-house developed analysis tools for the staphylococcal research community. The comprehensive set of genomic data in StaphyloBase will facilitate analyses on comparative genomics and pathogenomics among different staphylococcal strains or species. Although a related database on S. aureus, AureusDB (http://aureusdb.biologie.uni-greifswald.de), already exists, it has not been updated since 2007. Moreover, there are many differences between the AureusDB and our StaphyloBase. AureusDB was mainly designed to host the genome sequences of various S. aureus strains and related species, whereas StaphyloBase covers the genome sequences of strains and species under the whole Staphylococcus genus. In addition, StaphyloBase provides a set of useful analysis tools, particularly for comparative analysis, to analyse the staphylococcal genomic data. For example, StaphyloBase analysis is powered by two newly designed tools, namely, PGC for pairwise genome comparison and PathoProT for comparative pathogenomics analysis. The AJAX-based real-time search feature and JBrowse (11) have also been introduced in StaphyloBase to allow rapid and seamless searching and browsing of the staphylococcal genomes and annotations.
Database content and refinement
StaphyloBase is a central repository for the Staphylococcus genus that provides all the annotated information and data of ∼292 strains/genomes (Table 1) of at least 22 different species hosting 250 draft genomes and 42 complete genomes. The web interface enables users to execute quick, user-friendly and efficient browsing of strains with respect to their species and genome status. The ‘View Strains’ option in the Species table of the Browse page accessed from the home page provides significant features of genomes like genome size, G + C content, number of contigs, protein-coding sequences (CDS), tRNAs and rRNAs.
List of available staphylococcal strains/genome sequences in StaphyloBase
| Numbers | Species | Number of genomes | |
|---|---|---|---|
| Draft | Complete | ||
| 1 | Staphylococcus arlettae | 1 | 0 |
| 2 | Staphylococcus aureus | 160 | 33 |
| 3 | Staphylococcus capitis | 3 | 0 |
| 4 | Staphylococcus caparae | 1 | 0 |
| 5 | Staphylococcus carnosus | 0 | 1 |
| 6 | Staphylococcus delphini | 1 | 0 |
| 7 | Staphylococcus epidermidis | 61 | 2 |
| 8 | Staphylococcus equorum | 1 | 0 |
| 9 | Staphylococcus haemolyticus | 1 | 1 |
| 10 | Staphylococcus hominis | 4 | 0 |
| 11 | Staphylococcus intermedius | 1 | 0 |
| 12 | Staphylococcus lentus | 1 | 0 |
| 13 | Staphylococcus lugdunensis | 3 | 2 |
| 14 | Staphylococcus massilliensis | 2 | 0 |
| 15 | Staphylococcus pettenkoferi | 1 | 0 |
| 16 | Staphylococcus pseudintermedius | 0 | 2 |
| 17 | Staphylococcus saprophyticus | 1 | 1 |
| 18 | Staphylococcus simiae | 1 | 0 |
| 19 | Staphylococcus simulans | 1 | 0 |
| 20 | Staphylococcus sp. | 3 | 0 |
| 21 | Staphylococcus vintulinus | 1 | 0 |
| 22 | Staphylococcus warneri | 2 | 0 |
As S. aureus and S. epidermidis are the two best studied staphylococcal species, our StaphyloBase hosts the genomic data and annotations of 193 S. aureus and 63 S. epidermidis strains as well as 36 strains of other staphylococcal species. Annotations include open reading frame (ORF) type, functional classification, chromosomal position, nucleotide length, amino acid length, strand, subcellular localization, hydrophobicity and molecular weight. This information was generated by a combination of automated pipeline and manual curated steps. To make the genome annotation consistent and easier for comparative analyses across strains, we annotate all genomes of staphylococcal strains using RAST (Rapid Annotation using Subsystem Technology) (12). We automated this process by using Network-Based SEED (13) API modules with Perl scripts in submitting huge number of genome sequences and retrieving the annotated results from the RAST server. RAST is used to identify putative protein-coding genes, rRNA and tRNA genes. It also annotated the functions of the predicted genes by mapping these genes to subsystems. It should be noted that it could be a limitation in annotating these genomes using RAST approach only. However, we have created hyperlinks for ORFs, contigs and strain names, directing users to the GenBank sites where users can view the original GenBank annotations. StaphyloBase currently has a collection of 754 032 CDSs, 19 258 rRNA and 15 965 tRNAs from the 292 genomes of different staphylococcal species. PSORT (14) was used to determine subcellular localizations of each putative CDS. Prediction of subcellular localization is essential for giving insights into protein function and the identification of cell surface/secreted drug targets.
Real-time data searching feature
StaphyloBase hosts a huge amount of staphylococcal genomic data and annotation and this is expected to considerably increase as more genomes are sequenced in the future. Therefore, an interactive interface allowing users to rapidly search a large volume of genomic data is vital. To give the staphylococcal research community a user-friendly and seamless search experience, we implemented a powerful real-time AJAX-based search system in the ‘Search’ tool on the home page. Users can search for an ORF by using different parameters including species name, strain, ORF ID, keywords and type of sequence (Figure 1). Moreover, when users are keying the search keywords, the system will rapidly retrieve the matches from the StaphyloBase operating in real-time. This will help users to get the right keywords and will speed up the searching, both of which are crucial in searching a huge database.
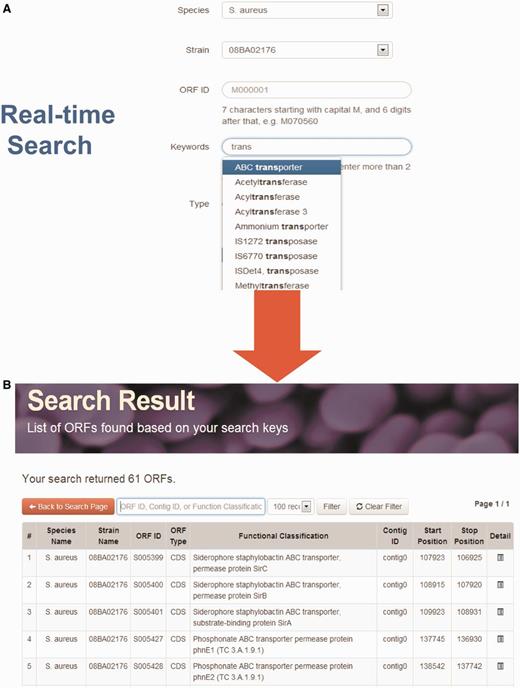
Real-time search function. (A) Users can search using different parameters. For example, they can search by keywords and a list of matches from the database will be displayed at the bottom in real-time. (B) Example of search output.
Tools and implementation
Pairwise Genome Comparison on the fly
StaphyloBase is not just designed as a genomic data repository, but also aims to be an analysis platform and is particularly designed to facilitate comparative analysis of the staphylococcal genomes. We have developed a Pairwise Genome Comparison (PGC) tool, which is an automated pipeline allowing users to determine the relationships between two closely related staphylococcal genomes (Figure 2 and Supplementary Figure S1). Through an input web interface on StaphyloBase, users can choose two genomes of interest in StaphyloBase for comparison. Alternatively, users can use an online custom web form to upload their own staphylococcal genome sequence for comparison with a staphylococcal genome in StaphyloBase. Different parameters like genome identity, link threshold (minimum length of aligned DNA fragments to be displayed) and merge threshold (limits the gap between two aligned DNA fragments where beyond the limit the two fragments are merged) can be set in the input web form. The influence of different parameters on the display of the aligned genomes with Circos (15) are shown in Figure 3. Once the job is submitted to our server, PGC will start aligning the two user-defined genomes using the NUCmer program in the MUMmer package (16). The output alignments from NUCmer will be processed and Circos (15) input files will be generated, using our in-house scripts, for visualizing the aligned genomes in a circular layout.
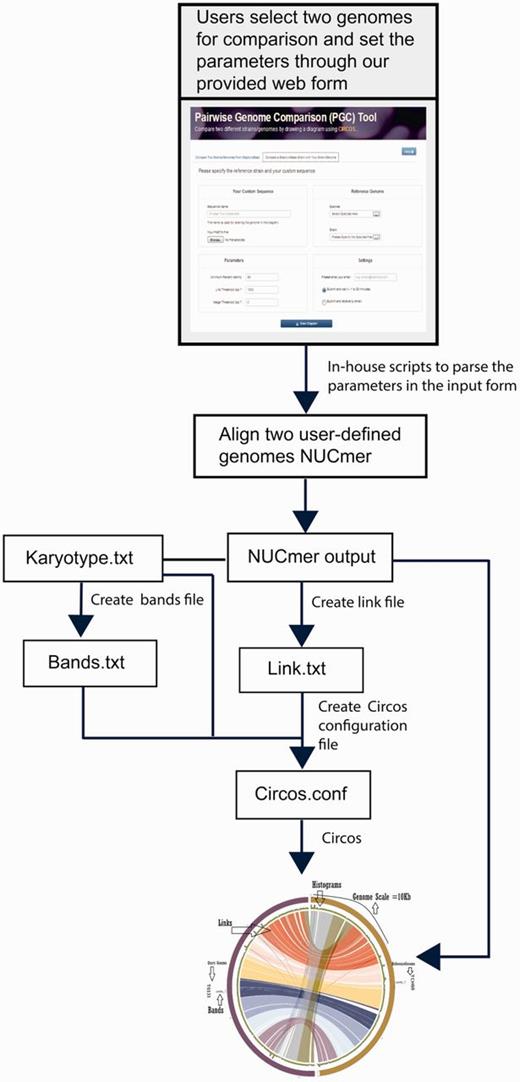
PGC workflow and automation for comparing two user-defined staphylococcal genomes. This includes the parsing of user-defined parameters and text files containing information, such as karyotype (in Circos, karyotypes are typically chromosomes or sequence contigs or clones in biological context), links, histograms and bands by Perl and Python scripts to create a Circos.conf file for displaying the aligned genomes with Circos.
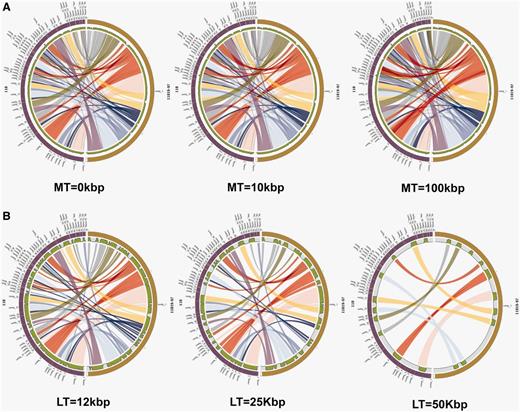
The influence of different cut-offs for the Merge Threshold (MT) and Link Threshold (LT). As a case study, the genomes of S. aureus strain 11819–97 and S. aureus strain 118 were compared using PGC tool. Each half circle (either left or right) represents each separate genome/assembly. The coloured links show the homologous regions in the two selected genomes. We can clearly observe how different user-defined thresholds affect the display of the two aligned genomes. (A) Different cut-offs for MT. Parameters: Genome Identity—95% and LT—1000 bp were used for all three plots. (B) Different cut-offs for LT. Parameters: Genome Identity—95% and MT—0 bp were used for all three plots.
Similar online visualization tools, such as Circoletto (17), ACT interface of IMG (18) and CoGe (19), have been developed and published for visualizing BLAST (20, 21) sequence comparison results of genomes. In fact, this tool works best with small datasets. However, there are many differences between PGC and other tools. One of the major differences is that other tools align sequences using BLAST (local alignment), whereas PGC uses NUCmer (global alignment), which is suitable for large-scale and rapid whole-genome alignment. For linear layout alignment tools (ACT interface of IMG and genome comparison tools in CoGe), it is difficult to organize links or relationships between two aligned genomes compared with the circular layout of PGC tool, which has advantage in visualizing the relationships in global view. Besides that, PGC allows users to adjust settings such as minimum percentage genome identity (%), merging of links according to merge threshold (bp) and the removal of links according to the user-defined link threshold (bp) through the provided online form. In the Circos plot generated by PGC, we have also added a histogram track showing the percentage of mapped regions along the genome (Supplementary Figure S1). This track is useful and helps users to identify putative indels and repetitive regions in the staphylococcal genomes.
Pathogenomics Profiling Tool
We have developed Pathogenomics Profiling Tool (PathoProT) to facilitate comparative pathogenomics analysis of the staphylococcal strains. The availability of genome sequences of different staphylococcal species enables comparative analyses of virulence factors in the staphylococcal pathogen genomes, which may provide new insights into pathogen evolution and the diverse virulence strategies used. Understanding the pathogenic mechanisms of these pathogens would aid the development of novel approaches for disease treatment and prevention.
PathoProT is specifically designed for the staphylococcal community to facilitate research on the pathogenicity of staphylococcal pathogens. Briefly, users can select a set of staphylococcal strains and parameters of interest (e.g. thresholds for sequence identity and completeness) for comparison using the online web form. This information is fed to the PathoProT pipeline that will initiate the prediction of the virulence genes in each selected staphylococcal strain by BLASTing the RAST-predicted CDSs against the manually curated virulence genes in the Virulence Factors Database (VFDB) (22–24). The putative virulence genes will be identified based on cut-offs set by the users. The output results are tabulated as data matrix and will be passed to R scripts for clustering; strains are clustered based on the virulence gene profiles, e.g. the presence and absence of virulence genes using hierarchical clustering algorithm (complete linkage method), followed by visualizing the end results as a heat map with dendrograms showing clustered strains with closely related sets of virulence genes, sorted according to similarities across the strains and genes (Figure 4).

Heat map showing clusters of the selected staphylococcal strains based on their virulence gene profiles. The columns represent the staphylococcal strains, whereas the rows represent the predicted virulence genes. A red box indicates the presence of the virulence gene in the corresponding strain. In contrast, a black box indicates absence. Through this clustering, it is easy to visualize the common virulence genes (e.g. nuc) across the selected staphylococcal strains/species, strain-specific virulence genes (e.g. sec, sel for strain S. epidermidis FR1909) and species-specific virulence genes (e.g. hysA, esaA essA, and essB are specific to all selected S. aureus).
This analysis tool can be used in several ways. For instance, users can identify the common as well as species- or strain-specific virulence genes through the generated heat map. Besides that, users can compare the virulence gene profiles of different groups of staphylococcal strains, e.g. non-pathogenic versus pathogenic strains. This may help to identify genes that are important for the pathogenicity of the pathogenic strains or investigate how a non-pathogenic strain has evolved into a pathogenic strain. Moreover, PathoProT will also cluster user-selected staphylococcal strains based on their virulence gene profiles, helping researchers to study the evolution of these strains and also to identify closely related strains/species based on their virulence gene profiles.
Homology Search Tools
BLAST (20) was implemented in the database to allow for easy homology searching for sequences of interest. StaphyloBase provides the BLAST function in the ‘Tools’ menu on the homepage. There are four different BLAST functions available: (i) BLASTN (compares nucleotide sequence against all RAST-predicted nucleotide gene sequences in StaphyloBase), (ii) BLASTN Whole Genome (compares nucleotide sequence against all staphylococcal genome sequences in StaphyloBase), (iii) BLASTP (compares protein sequence against all RAST-predicted protein sequences in StaphyloBase) and (iv) BLASTX (compares the six-frame conceptual translation products of a nucleotide query sequence (both strands) against all RAST-predicted protein sequences in StaphyloBase. The results are sorted by alignment scores. In addition, BLAST VFDB is also incorporated into StaphyloBase, allowing users to examine whether their sequences of interest are virulence genes based on homology search against VFDB (22–24).
Interactive AJAX-based genome browser
Many genome browsers have recently been created, each with its own strengths and weaknesses. We have chosen JBrowse for StaphyloBase for two main reasons: (i) most of the traditional genome browsers, for example, GBrowse (25), are implemented using the Common Gateway Interface (CGI) protocol. Using these CGI-based genome browsers, the whole-genome browser page needs to be reloaded when users change how the data are displayed, which incurs a delay in response and negatively affects the user experience and (ii) with the advances in next-generation sequencing technologies and bioinformatic tools, we anticipate that many more staphylococcal genomes will be sequenced and annotated. Therefore, a user-friendly genome browser that allows rapid and seamless browsing of high volumes of genomic data will be a major advantage.
To give users a seamless browsing experience, we incorporated AJAX-based JBrowse (11) into StaphyloBase. Using this next-generation genome browser, users can navigate the staphylococcal sequence and annotation data over the web and this helps preserve the user’s sense of location by avoiding discontinuous transitions, offering smooth animated panning, zooming, navigation and track selection. The user can easily browse a genomic region in the provided search box in the JBrowse window (Figure 5). Besides that, each track (e.g. CDS, DNA or RNA tracks) can be easily turned on/off or customized by clicking on it. All displayed features (e.g. CDSs and RNAs) are clickable and will link to a window showing detailed information about the selected feature. An example of the visualization of the annotated features of a genomic region in JBrowse is shown in Figure 5.
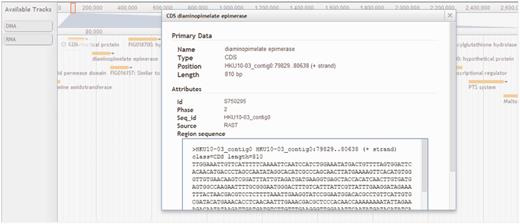
Browser panel showing a genomic region associated with a gene encoding diaminopimelate epimerase and its neighbouring genes. CDS, DNA and RNA are shown in different tracks.
Database development
The web interfaces were developed in PHP-HTML5 using CodeIgniter and Twitter Bootstrap as back-end and front-end frameworks, respectively. The MySQL relational database was used to store genomic annotations generated from published software and in-house scripts in this project. A specific naming convention was used to uniquely identify each ORF in StaphyloBase; each ORF ID starts with S as suffix followed by six digits (e.g. S545817). The download page provides options for users to download genomic sequences and annotations according to species name, strain name and type of data. The types of data that users can download are genome sequences, ORF annotation table (csv format), CDS sequences, RNA sequences and ORF sequences.
Discussion
With advances in high-throughput sequencing technologies, it is imperative that the abundant data generated can be easily accessible for analysis. With StaphyloBase, we aim to provide a one-stop resource platform that will make it easy to access and analyse whole-genome genomic data and information for staphylococcal bacteria through an organized and user-friendly interface. PGC and PathoProT are some of the bioinformatics tools for the comparative analysis implemented in StaphyloBase, which allow researchers to conveniently assimilate and explore the data in an intuitive manner.
The increasing availability of new genome sequences in the future will require continuous updates to StaphyloBase and more analysis tools will be added, allowing researchers to further analyse the genomic data of the staphylococcal bacteria. To progressively enhance StaphyloBase, we highly encourage the scientific community to send us feedback and suggestions on improving this database; sharing curated data and requests for additional functions or analysis tools are most welcome. We hope that this project will be useful to the community, providing the wealth of genome information in a single repository integrated with an analysis platform facilitating future studies of the staphylococcal bacteria.
Acknowledgements
The authors would like to thank Dr Robert White (Department of Physiology, Development and Neuroscience, University of Cambridge) for his assistance in proofreading the manuscript. They also thank all members of the Genome Informatics Research Group (G.I.R.G) in contributing their efforts in improvement of StaphyloBase.
Funding
This work was funded by Ministry of Education & University of Malaya (grant number: UM.C/625/1/HIR/MoHE/08). Funding for open access charge: UM.C/625/1/HIR/MoHE/08.
Conflict of interest. None declared.
References
Author notes
†These authors contributed equally to this work.
Citation details: Heydari,H., Mutha,N.V.R., Imran Mahmud,M., et al. StaphyloBase: a specialized genomic resource for the staphylococcal research community. Database (2014) Vol. 2014: article ID bau010; doi:10.1093/database/bau010.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}