





Abstract
Next generation sequencing (NGS) innovations put a compelling landmark in life science and changed the direction of research in clinical oncology with its productivity to diagnose and treat cancer. The aim of our portal comprehensive resources for cancer NGS data analysis (CRCDA) is to provide a collection of different NGS tools and pipelines under diverse classes with cancer pathways and databases and furthermore, literature information from PubMed. The literature data was constrained to 18 most common cancer types such as breast cancer, colon cancer and other cancers that exhibit in worldwide population. NGS-cancer tools for the convenience have been categorized into cancer genomics, cancer transcriptomics, cancer epigenomics, quality control and visualization. Pipelines for variant detection, quality control and data analysis were listed to provide out-of-the box solution for NGS data analysis, which may help researchers to overcome challenges in selecting and configuring individual tools for analysing exome, whole genome and transcriptome data. An extensive search page was developed that can be queried by using (i) type of data [literature, gene data and sequence read archive (SRA) data] and (ii) type of cancer (selected based on global incidence and accessibility of data). For each category of analysis, variety of tools are available and the biggest challenge is in searching and using the right tool for the right application. The objective of the work is collecting tools in each category available at various places and arranging the tools and other data in a simple and user-friendly manner for biologists and oncologists to find information easier. To the best of our knowledge, we have collected and presented a comprehensive package of most of the resources available in cancer for NGS data analysis. Given these factors, we believe that this website will be an useful resource to the NGS research community working on cancer.
Database URL : http://bioinfo.au-kbc.org.in/ngs/ngshome.html .
Introduction
The chain termination method by Sanger and sequencing method by Maxam-Gilbert overturned the biomedical world through an efficient sequencing approach at significantly lower costs ( 1 , 2 ). In 2004, 454 Life Sciences showcased a paralleled form of sequencing called pyrosequencing ( 3 ). The first form of their instrument decreased sequencing expenses at 6-fold contrasted with mechanized Sanger sequencing, and was the second of the new era of sequencing innovations, after massive parallel signature sequencing ( 4 ). The main difference between Sanger sequencing data and next generation sequencing (NGS) data is the read length or the quantity of nucleotides acquired. NGS is a recent innovation that empowers massively parallel sequencing reactions along these lines diminishing the specimen size and reagent costs. The sequencing process manifold to permit concurrent sequencing each reaction and to analyse the huge number of samples. Procedures in NGS include extracting DNA/RNA from samples, making a library of sections that are sequenced in parallel to short reads, and are reassembled by aligning them to a reference genome. In this way, the entire genome is obtained from the arrangement of consensus reads. NGS utilizes different platforms such as GS FLX by 454 Life Technologies/Roche, Genome Analyzer by Solexa/Illumina, SOLiD by Applied Biosystems, CGA Platform by Complete Genomics, PacBio RS by Pacific Biosciences, Polonator G.007, Ion/Proton PGM and Oxford Nanopore for sequencing genomes ( 5 ). The reads obtained from these platforms can be aligned and further analysed by using various NGS tools.
NGS experiments generate volumes of data, which requires a computationally intensive system for data storage, management and processing. The main processing feature of the system is to transform image data into sequence reads, known as base calling. On each platform, for each base in reads, image parameters such as intensity level, background and noise are utilized to generate reads and quality scores. Quality scores computed provides significant information for downstream analysis. Assembly and alignment are considered to be complicated and resource intensive steps in the NGS data analysis. The RNA data analysis also puts forward unique challenges and demands sequence alignment across spliced junctions and differential expression. In addition to that, variant calling for analysing variants, annotation for adding biological context, ChIP sequencing and methylation for analysing gene regulation are special tasks in NGS data analysis. Major applications of NGS are detecting genomic alterations and biomarkers which in turn be useful in diagnosis and treatment of cancer.
Cancer is an array of diseases defined by abnormal cell growth and is caused by mutations in somatic or germ-line cells. NGS technologies play a critical part in the diagnosis and treatment of cancer. Researchers are using NGS technologies to achieve a deeper understanding of tumor through target sequencing and to study cancer progression. Emerging methods in NGS are useful in monitoring the progression of cancer and drug response in cancer cells. Various NGS tools have been developed to analyse and interpret sequence reads and different studies have been carried out to find novel genomic variations, which cause cancer. NGS based studies through whole genome sequencing (WGS) and whole exome sequencing (WES) technologies will help us to understand the mechanism underlying progression and evolution of cancer ( 6 ).
The exponential growth of NGS data with extensive cancer studies and the development of new tools made easier for the research community to analyse NGS data. The primary bottleneck of NGS study lies in data analysis, because the complexity of NGS data analysis depends on multitude of databases, tools and heterogeneity of data involved in the study. The data analysis workflow needs to be designed carefully and tools have to be selected cautiously for structured data management and meaningful biological results. The NGS tools can be classified into commercial packages and open source tools. Commercial packages available are DNANexus, CLC Genomics Workbench and Genome Quest. Most commercial packages use proprietary algorithms for data analysis and are costlier. In contrast, researchers developed excellent software tools that may be either standalone or web-based for the analysis of progressively large genome data and made these tools open access to all.
Methods and Resources
NGS tools for data analysis
NGS tools used in our web portal are grouped into five categories. They are cancer genomics, cancer transcriptomics, quality control (QC), cancer epigenomics and cancer genome visualization. The workflow of NGS tools is shown in Figure 1 . Different tools listed are restricted to the criteria that they are available for either online or standalone and are strictly confined to analyse NGS data.
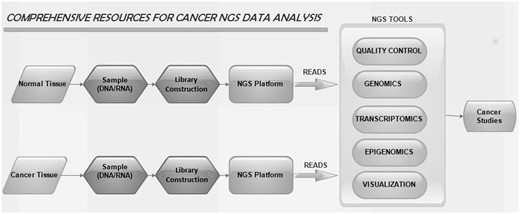
The workflow of NGS tools in cancer studies.
Cancer genomics
The term ‘Genomics’ was first coined by McKusick and Ruddle in September 1987 ( 7 ) as a name for their new journal. Genomics is a discipline that applies to analysing the structure and function of genomes. Cancer genomics denote sequencing a genome that is confined to a particular tumor tissue and then mapping the short reads obtained to a reference genome. Cancer genomics tools were clustered into four main groups such as assembly, alignment, annotation and variation discovery based on their application. Variation discovery tools under distinct subsections like mutation calling, structural variations, copy number variations (CNVs), indel and mutation effects are listed along with other cancer genomics tools for annotation, assembly and aligment in Table 1 . Assembly denotes aligning and blending short reads from NGS experiment to recreate the original sequence. Alignment is a crucial step in resequencing and it refers to align short reads from NGS experiment to a reference genome. With reference genome in hand, alignment is used for detecting variants in samples. Annotation is the process of identifying the position and biological context of genes in a genome. Genome variations include mutations, polymorphisms and structural variations. The term mutation is often used to refer to a genome variation that is related to certain hereditary disease or cancer, while polymorphism indicates a modification that can be neither harmful nor beneficial ( 65 ). Various tools have been listed under genomic variation discovery. They are grouped into five subdivisions such as mutation calling, structural variations, CNVs, indel and mutation effects. Mutation calling tools are used to identify and annotate mutations in reads. Indel tools are used to find small indels, and structural variation tools are used to detect changes in chromosomal structure. CNVs are genomic differences that occur due to deletion or duplication of larger regions of DNA. Moreover, the effect of mutation on a specific site occurred by certain amino acid substitution can be predicted by using mutation effect predication tools. Even though the tools are listed under precise section, they may serve more than one purpose. For example, VarScan tool though listed under CNV, also detect indels.
List of tools for cancer genomics
Cancer transcriptomics
Transcriptomics is the study of complete RNA transcript (transcriptome) produced by a genome at specific conditions. NGS technologies are applied to study cDNA fragments to deliver a transcriptional profile. Transcriptomics involves alignment and analysis of RNA sequence reads and they are aligned using RNA specific aligners to detect new splicing junctions. Differential expression tools are used to quantify the expression values of reads. Gene fusion tools are used to align reads comprising fusion junctions to the genome. Technical improvements and decreasing expenses made transcriptome analysis a routine in cancer research and it provides boundless potential in cancer research. The transcriptomic tools are classified as spliced alignment, differential expression, alternative splicing and gene fusion and are listed in Table 2 . They are used to understand how transcripts are altered by diseases such as cancer and how these altered transcripts play a significant role in distinguishing cancer and its subtypes ( 88 ).
List of tools for cancer transcriptomics
Quality control
QC is the first step in the NGS data analysis after getting raw sequence reads from next generation sequencers. In NGS experiments, shorter reads obtained may contain erroneous data like poor quality reads, adapter sequences, base calling errors and some insertions/deletions among the original reads. Definite screening techniques and filtration criteria like sequence quality, sequence length, etc. are used to minimize errors in sequence reads ( 89 ). In addition to these methods, certain software tools are used to detect contaminated and low quality reads called QC tools. QC tools use different algorithms to detect and filter artifacts in reads obtained from NGS methods. The error detection and correction tools for QC are listed in Table 3 . The reads obtained after QC are further filtered for primer contamination to improve and ensure quality of reads. Read quality has to be checked carefully before initiating NGS data analysis because there is no utility present in downstream analysis tools to remove erroneous data in reads. In short, the quality of the output depends on the quality of the input in terms of quality reads.
List of tools for QC
Cancer epigenomics
Many cancers involve multiple factors like environmental factors or genetic factors with impact on interlinked biological pathways and the environmental effects are mediated through epigenetic modifications. The study of epigenetic changes that occur on a genome is referred as epigenomics. The advent of NGS, has empowered significant progress in the study of triggering, and progression of cancer. Epigenetic changes such as DNA methylation, modification of histones and miRNA silencing are also responsible for cancer. However, they do not produce any nucleotide change in the sequence. DNA methylation, histone modifications and furthermore, miRNA silencing play a major role in gene regulation. Sometimes, loss of methylation at general methylated sites (hypomethylation) and gain of methylation at the abnormal sites (hypermethylation) lead to cancer ( 105 ). ChIP Seq tools are used to discover motifs and identify histone modifications from enriched domains and peak regions. Epigenetic changes in a genome have the potential to explain complex disease mechanisms. In particular, DNA methylation plays a major role in genome evolution and histone modification. Methylation tools are used to generate methylation maps for analysis. Different available tools for cancer epigenomics are classified as Methylation, ChIP Seq and Bisulphite Seq, and they are listed in Table 4 .
List of tools for cancer epigenomics
Cancer genome visualization
The alignment and assembly data can be examined by using graphical tools for analysing the output files such as FASTQ, SAM (Sequence Alignment Map format), BAM (Binary compressed SAM format), VCF (Variant Call Format), etc. from various NGS tools. Genome visualization tools provide an interface to visualize data, results and annotations associated with a particular genome of interest. Annotation data, genetic information, transcripts pattern, etc. are provided along with the genomic data. The visualization tool can either be a standalone tool that can be installed on a local computer or a web browser tool. Most visualization tools are provided with a graphical user interface (GUI) so that user can view data or results, edit data, color and zoom. In some tools, search operations can also be performed. Visualization tools for data visualization with data interpretation are listed in Table 5 .
List of tools for visualization
| Category | Tool | URL | Reference |
|---|---|---|---|
| Visualization | Strand NGS | http://www.strand-ngs.com/ | |
| CIRCOS | http://circos.ca/ | ( 123 ) | |
| IGV | http://www.broadinstitute.org/igv/ | ( 124 , 125 ) | |
| Tablet | http://ics.hutton.ac.uk/tablet | ( 126 ) | |
| BamView | http://bamview.sourceforge.net/ | ( 127 , 128 ) | |
| EagleView | http://bioinformatics.bc.edu/marthlab/wiki/index.php/EagleView | ( 129 ) | |
| NGSView | http://ngsview.sourceforge.net/ | ( 130 ) | |
| ZOOM Lite | http://bioinfor.com/zoom/lite | ( 131 ) | |
| UCSC Genome Browser | http://genome.ucsc.edu/ | ( 132 ) | |
| Genplay | http://genplay.einstein.yu.edu/wiki/index.php/Main_Page | ( 133 , 134 ) | |
| Savant | http://genomesavant.com/p/savant/index | ||
| ABrowse | http://www.abrowse.org/ | ( 135 ) | |
| Integrated Genomic Browser | http://bioviz.org/igb | ||
| Artemis | http://www.sanger.ac.uk/resources/software/artemis | ( 136 , 137 ) |
NGS pipeline tools
Many tools are available for NGS data analysis, yet their use often limited to skilled bioinformaticians since these tools have been developed in different programming languages for different operating systems. For instance, Bowtie is an excellent tool for aligning sequencing reads but will be complicated for biologists to install, configure and use. To overcome the difficulty of individual tool developers designed certain workflows called pipelines. Managing NGS reads, handling and configuring NGS tools are difficult tasks for biologists and biotechnologists who work on NGS data. NGS Pipelines, a collection of structured commands or software tools specific to a particular platform or data are used to improve productivity and specificity of data processing. Pipelines can be either general (for data analysis) or specific (for QC and variation calling). Pipelines implement simple user interface, and most of the tools are cross platform ( 138 ). Variant calling pipeline tools are used to detect aberrations, polymorphisms and indels. Variant calling pipeline tools, QC pipelines and data analysis pipelines are listed in Table 6 . Recent development in pipelines and protocols permit researchers to overcome the technical issues related to handling NGS tools. For instance, in Galaxy webserver ( https://usegalaxy.org/ ), pipelines are referred as customized workflows which include more than one Galaxy tool in sequential form for automated running of tools. Another example of pipeline is DDBJ read annotation pipeline, which is a cloud based pipeline for annotation of NGS data reads. The DDBJ Pipeline offers a GUI for processing NGS datasets using decentralized processing by NIG supercomputers currently at free of cost ( 140 ). The success of NGS data analysis lies in the selection of NGS pipeline specific to particular NGS platform and organism of study.
List of pipelines
NGS file converters
Most common file formats related to NGS data analysis are FASTA, FASTQ, QSEQ, SFF (Standard Flowgram Format), SAM, BAM, VCF, BED (Browser Extensible Data format), etc. Most NGS sequence files are in FASTQ or FASTA formats, which incorporate reads and quality scores. If sequence reads are mapped to the reference sequence, we get either SAM or BAM file format as output files. Sometimes it might be vital to convert one file format to another for data analysis. For instance, VCF with gene sequence variation information is no longer maintained by the 1000 Genomes Project ( http://www.1000genomes.org/ ) and QSEQ files are plain text files generated by earlier Illumina machines. So, we need to convert these file formats into commonly used file formats like FASTQ for analysis ( 149 , 150 ). The tools used for NGS file format conversion are listed in Table 7 .
List of tools for file format conversion
| Category | Tool | URL | Reference |
|---|---|---|---|
| File converters | SRA Toolkit | http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=show&f=software&m=software&s=software | |
| FASTX-Toolkit | http://hannonlab.cshl.edu/fastx_toolkit/ | ||
| NGSQC Toolkit | http://www.nipgr.res.in/ngsqctoolkit.html | ( 89 ) | |
| Picard | http://broadinstitute.github.io/picard/ | ||
| Bamtools | https://github.com/pezmaster31/bamtools | ||
| SAMtools | http://samtools.sourceforge.net/ | ||
| GenePattern | http://www.broadinstitute.org/cancer/software/genepattern/modules?taskType=Data+Format+Conversion | ||
| PRINSEQ | http://prinseq.sourceforge.net/ | ||
| PGDSpider | http://www.cmpg.unibe.ch/software/PGDSpider/ | ||
| Galaxy | https://usegalaxy.org/ |
Cancer resources
Cancer resources, although not mainly useful to individual patients, are essential for healthcare professionals and researchers to develop strategies that can tackle challenges posed by cancer. Among the resources available for cancer, The Cancer Genome Atlas (TCGA) Data Portal furnishes an important platform for researchers to download, and analyse data sets generated by TCGA ( 151 ). Cancer resources section contains four different types of data which might be useful to any researcher working with cancer. They are (i) Cancer study data, list of articles clustered under different cancer types. (ii) Cancer Databases, list of cancer databases and oncogenomic browsers available. (iii) Cancer projects, list of ongoing projects in cancer. (iv) Cancer Pathways, list of cancer pathways. Meta analysis is a statistical analysis that is connected to comparative experiments of different and independent researchers that includes pooling the data and utilizing the pooled information to test the effectiveness of the study ( 152 ). In cancer data resources, literature data have been collected and included in the list only if the study was on cancer oriented in human and method of sequencing used must be NGS and also the literature must be published in peer-reviewed journals. The collected list of literature is displayed in the form of a list. Under cancer databases section, browsers and databases listed provide cancer related information like oncogenes, suppressor genes, methylation data and mutation data. In cancer projects section, different cancer projects by research centers and Institutes like Wellcome Trust ( http://www.sanger.ac.uk/research/projects/cancergenome/ ), International Cancer Genome Consortium ( https://icgc.org/icgc ), etc. are incorporated to understand the molecular basis of cancer and gene expression profiles of different cancer types at different stages. In Cancer Pathways section, interactive pathway maps of different types of cancer from KEGG PATHWAY ( www.genome.jp/kegg/ ) database are listed. The interactive pathway map helps us to understand interrelated oncogenes for each cancer listed.
Web page development
The web pages were developed using hyper text markup language (HTML) language and cascaded style sheets (CSSs) for consistent styling with hyperlinks to various tools, literature, databases, pathways and projects.
Database construction
The 1000 Genomes Project was the first multi-terabytes submitter to two sequence read archives (SRAs), the European nucleotide archive (ENA) SRA and the NCBI SRA ( 153 ). SRA data from NGS platforms make sequence data access to researchers to enhance reproducibility and novel discoveries by analysing data sets. The literature data and SRA data extracted from the NCBI SRA ( http://www.ncbi.nlm.nih.gov/sra/ ) were stored using MySQL ( http://www.mysql.com/ ), an extensively used open source relational database management system for biological research. The literature data collected from NCBI PubMed ( http://www.ncbi.nlm.nih.gov/pubmed/ ), were annotated with gene data so that the literature search could be done based on either cancer or gene. The literature data include all primary citation details like Author, Title, PubMed ID (identifier), Cancer type and Journal Details. Literature and gene data include gene id in addition to all primary citation details. SRA data listed in the table consist of experiment accession, study accession, title, submitter, technology, library source and library selection.
Search page implementation
Comprehensive resources for cancer NGS data analysis (CRCDA) can be queried based on (i) type of data and (ii) type of cancer. The data available for search is of three types, (i) literature and gene data, (ii) literature data and (iii) SRA data. The literature and SRA data can be queried using the search page and the search scripts were coded using PHP, a widely used scripting language. The literature database contains articles related to major cancer types such as lung, liver, breast, colorectal, prostate, gastric, cervix, bladder, non-Hodgkin lymphoma, leukemia, pancreas, kidney, endometrial, oral, thyroid, brain, ovary and skin cancers. Cancer types were selected based on their abundant existence in world population. SRA data for certain cancer types like esophagal and prostate cancer was not available at the time of database construction. So, SRA data for these types of cancer will be uploaded into the database later. The literature data can be queried either based on cancer type or gene name. The search page for literature can be accessed in two ways as shown in the following Figures 2 and 3 . For example, in Figure 2 the literature data for breast cancer can be searched by selecting ‘breast cancer’ in cancer type from dropdown menu ( Figure 2 ) and in Figure 3 the literature data for gene ‘BRCA1’ can be selected by selecting BRCA1 from gene dropdown menu. The literature data listed in gene data include all cancer types which involve BRCA1 ( Figure 3 ). The literature data were listed as default from January 1995 to December 2014. So, user can select data from any time period within this specified limit, and the user can also search the database using the first author’s name and journal details.
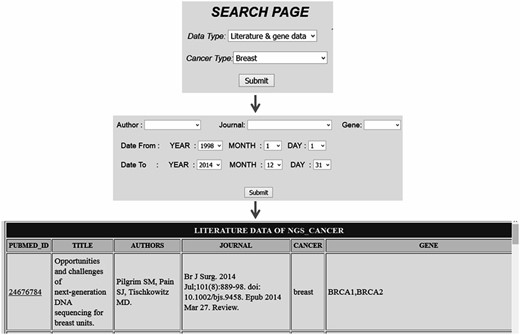
Search page accessed based on cancer type which lists all citation details with gene data for a particular cancer type.
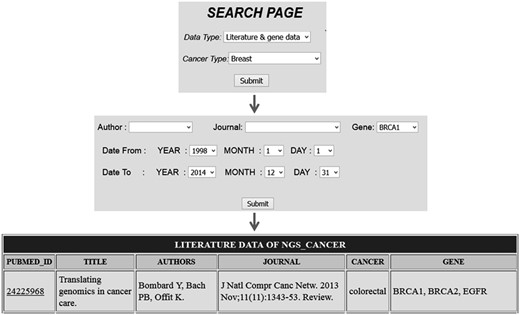
Search based on gene name which list all citation details for a particular gene in all cancer types.
Conclusion
The main application of NGS technology through WES and WGS in cancer research has made researchers to understand the molecular landscape of different types of cancer. CRCDA is the first web portal which provides literature, tools, pipelines, pathway and SRA specific to NGS and cancer. Here, we have listed nearly 180 and above software tools in the portal under tools and pipelines and more than 500 publication information of NGS studies, which would be useful for researchers working in oncology. Peer-reviewed articles on NGS-cancer studies, cancer databases, cancer pathway data would also be beneficial to enrich cancer research in a more efficient way. Availability of all cancers and NGS-related information in one portal provides very easy and quick reference for oncology researchers.
Future Work
Future plans include updating tools and literature data once in every 6 months to remove outdated tools and to update literature data in the database. A search page has been planned to search tools under each category and a rating option to help users to select and use most rated best tools. Public data mining tools will also be incorporated to enhance the value of this database.
Acknowledgements
The authors are greatly indebted to PhD Student Nupoor Chowdhary for her support in critical reading of the article.
Conflict of interest . None declared.
References
Author notes
Citation details: Manonanthini,T. and Ramesh Kumar,G. CRCDA—Comprehensive resources for cancer NGS data analysis. Database (2015) Vol. 2015: article ID bav092; doi:10.1093/database/bav092

{kind=link}
{kind=link}
{kind=link}