





Abstract
Hypersplenism in adults with homozygous sickle cell disease (HbSS) is rare, as most patients undergo autosplenectomy during early childhood. However, phenotypic variants such as high fetal hemoglobin, coexisting hemoglobinopathies, and different globin gene haplotypes can lead to splenic preservation and altered disease progression. These patients often exhibit milder symptoms, delayed complications, and increased morbidity. We report a rare case of a 43-year-old female with HbSS and elevated fetal hemoglobin who presented with hypersplenism, characterized by massive splenomegaly, pancytopenia, and compensatory bone marrow proliferation. She underwent an open splenectomy with an uneventful postoperative course. This case highlights the importance of recognizing atypical presentations in adult patients with sickle cell disease, in whom splenomegaly may persist. Clinicians should maintain a high index of suspicion to facilitate timely diagnosis and appropriate management, which can significantly improve patient outcomes.
Introduction
The natural course of splenic manifestations in patients with homozygous sickle cell (HbSS) disease entails temporary and mild splenomegaly in early childhood. Over time, recurrent vaso-occlusive events and splenic infarctions lead to progressive fibrosis and loss of splenic function [1]. This results in auto splenectomy or functional asplenia by the age of 6 years in the majority of these patients. Hence, hypersplenism, a condition characterized by a triad of splenomegaly, cytopenias, and compensatory bone marrow proliferation, is extremely rare in HbSS patients [2]. However, HbSS patients with high levels of fetal hemoglobin (HbF) are found to have less severe and delayed clinical manifestations and complications, with preservation of splenic function [3]. Splenic preservation till adulthood can lead to complications such as hypersplenism, predisposing these patients to increased morbidity. Here, we describe a case of a 43-year-old female with HbSS disease who presented with hypersplenism and massive splenomegaly, requiring splenectomy.
Case presentation
A 43-year-old female, a known case of sickle cell disease, presented with a 1-year history of epigastric pain and yellowish discolouration of sclera. The pain was insidious in onset, mild to moderate in intensity, continuous, dull aching, lasting 15–30 minutes per episode, and associated with exertional shortness of breath. She had a past history of HCV infection that was successfully treated, with a current viral load <20 IU/ml and undetectable HCV RNA. She denied smoking and alcohol use. There was no significant surgical or family history, though she had a history of multiple blood transfusions. She was on hydroxyurea 500 mg and folic acid 5 mg.
On examination, she was normotensive (100/65 mmHg), tachycardic (140 bpm), and febrile (38°C), with a respiratory rate of 21 breaths/min. Abdominal examination revealed mild epigastric tenderness and a palpable enlarged spleen. There were no features of acute chest syndrome or other systemic abnormalities. Laboratory investigation was consistent with anemia (Hb: 8.4 gm/dl, hematocrit: 32%) and thrombocytopenia (platelet: 97000 /mm3) with mild leukopenia (WBC: 3900 /mm3). Hemoglobin electrophoresis confirmed HbSS disease pattern with high HbF (HbA2: 2.5%, HbF: 19%, and HbS: 78.5%). The Direct Coombs test was negative. Peripheral blood smear showed normocytic, normochromic RBCs with mild polychromasia, ovalocytes, and target cells. Bone marrow aspiration revealed hypercellular marrow with significant erythroid hyperplasia and adequate iron stores. The liver function test revealed increased total (3.0 mg/dl) and direct (1.5 mg/dl) bilirubin. Her serum protein and albumin levels were 2.8 gm% and 1.0 gm%, respectively.
Abdominal ultrasonography revealed splenomegaly, with the spleen measuring approximately 25 cm, and a distended gallbladder with multiple pigmented gallstones. A diagnosis of HbSS disease with hypersplenism with cholelithiasis was made. After hemato-oncology consultation, the patient underwent open splenectomy and cholecystectomy. The spleen was massively enlarged, with a dimension of 25 × 15 cm2 (Fig. 1), and appeared congested, with multiple collateral vessels and a distributive pattern of the splenic artery. The procedure was uneventful with an estimated blood loss of 1000 ml. Postoperatively, she received two units of packed RBCs with supportive hydration and oxygenation. Her hematological parameters improved after a week with hemoglobin: 9.5 gm/dl, WBC: 11 000 / mm3, and platelets: 3 25 000 / mm3. She was discharged on hydroxyurea and folic acid and remained well at 6-month follow-up.
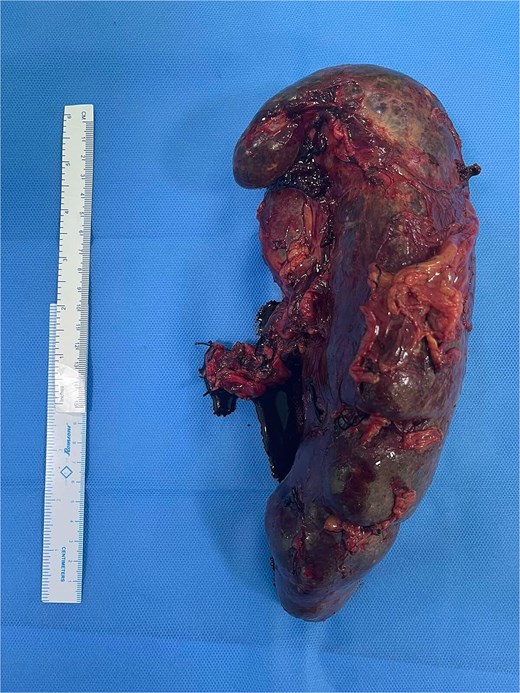
Gross specimen showing a massively enlarged, congested spleen measuring 25 × 15 cm2.
Discussion
Sickle cell disease (SCD) is a hereditary hemoglobinopathy caused by a point mutation (glutamate to valine) in the β-globin gene, leading to the formation of HbS instead of normal HbA. Under hypoxic conditions, HbS polymerizes, resulting in red blood cell sickling. Recurrent sickling causes repeated tissue infarction, leading to manifestations such as hemolytic anemia, vaso-occlusive crises, and acute chest syndrome [4]. In most HbSS patients, recurrent splenic infarctions from sickled RBCs lead to progressive fibrosis and functional loss by around 6 years of age, resulting in autosplenectomy [2]. Consequently, splenomegaly and hypersplenism are uncommon in these patients [5]. However, phenotypic variability in SCD may result in milder, delayed manifestations with preservation of splenic function into adulthood.
Phenotypic variability in SCD is influenced by factors such as elevated HbF, co-inherited hemoglobinopathies, and β-globin haplotypes [4]. HbF inhibits HbS polymerization under hypoxic conditions, reducing red cell sickling and resulting in fewer crises, milder complications, and delayed autosplenectomy [3]. HbF levels may be elevated in conditions such as hereditary persistence of fetal hemoglobin, anemia, bone marrow disorders, thyrotoxicosis, pregnancy, and with hydroxyurea therapy [4]. Coexisting hemoglobinopathies (e.g. α-thalassemia, β- and δβ-thalassemia, HbC, HbSC, Hb D-Punjab, and Hb O-Arab) can further attenuate disease severity [4]. The Arab-Indian β-globin haplotype is associated with higher HbF levels (~20%) [6]. Given our patient’s geographic background and HbF level of 19%, this variant may have contributed to preservation of splenic function into the fourth decade.
Preserved splenic function offers protection against severe infections through continued antigen processing and antibody production [7]. However, it may also predispose to complications such as acute splenic sequestration, splenic abscess, and hypersplenism [2]. Hypersplenism involves chronic sequestration of blood cells, resulting in cytopenias, splenomegaly, and compensatory bone marrow proliferation. Intrinsic splenic disorders lead to primary hypersplenism while hemoglobinopathies like SCD result in secondary hypersplenism [8]. In our case, the patient had pancytopenia (anemia, thrombocytopenia, and leukopenia), massive splenomegaly and hypercellular bone marrow, along with episodic epigastric pain and jaundice secondary to hemolysis.
Splenectomy, though not curative, is a widely practiced treatment for splenomegaly with secondary hypersplenism and forms part of long-term management in SCD. Indications include hypersplenism, recurrent severe splenic sequestration crisis, splenic infarction, intractable pain, and splenic infections complicated by abscess. In cases of massive splenomegaly, splenectomy may be associated with significant intraoperative blood loss. To reduce this risk, adjunctive partial splenic artery embolization (PSAE) can be performed prior to surgery [9, 10]. However, PSAE is associated with complications such as post-embolization syndrome, infections, and portal vein thrombosis [11].
Conclusion
In conclusion, splenomegaly with hypersplenism in adults with HbSS disease is rare and may reflect variable disease severity. This case highlights the need for a high index of suspicion and timely, multidisciplinary management to improve outcomes. Larger population-based studies are warranted to better define its epidemiology and the role of phenotypic variation in disease pathogenesis and prognosis.
Acknowledgements
Special thanks to the nurses, staffs and doctors who were directly or indirectly involved in the care of the patient.
Conflicts of interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Funding
None declared.
Ethical approval
Ethical approval by the ethics committee is not required for a case report in our country.
Informed consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.

{kind=link}